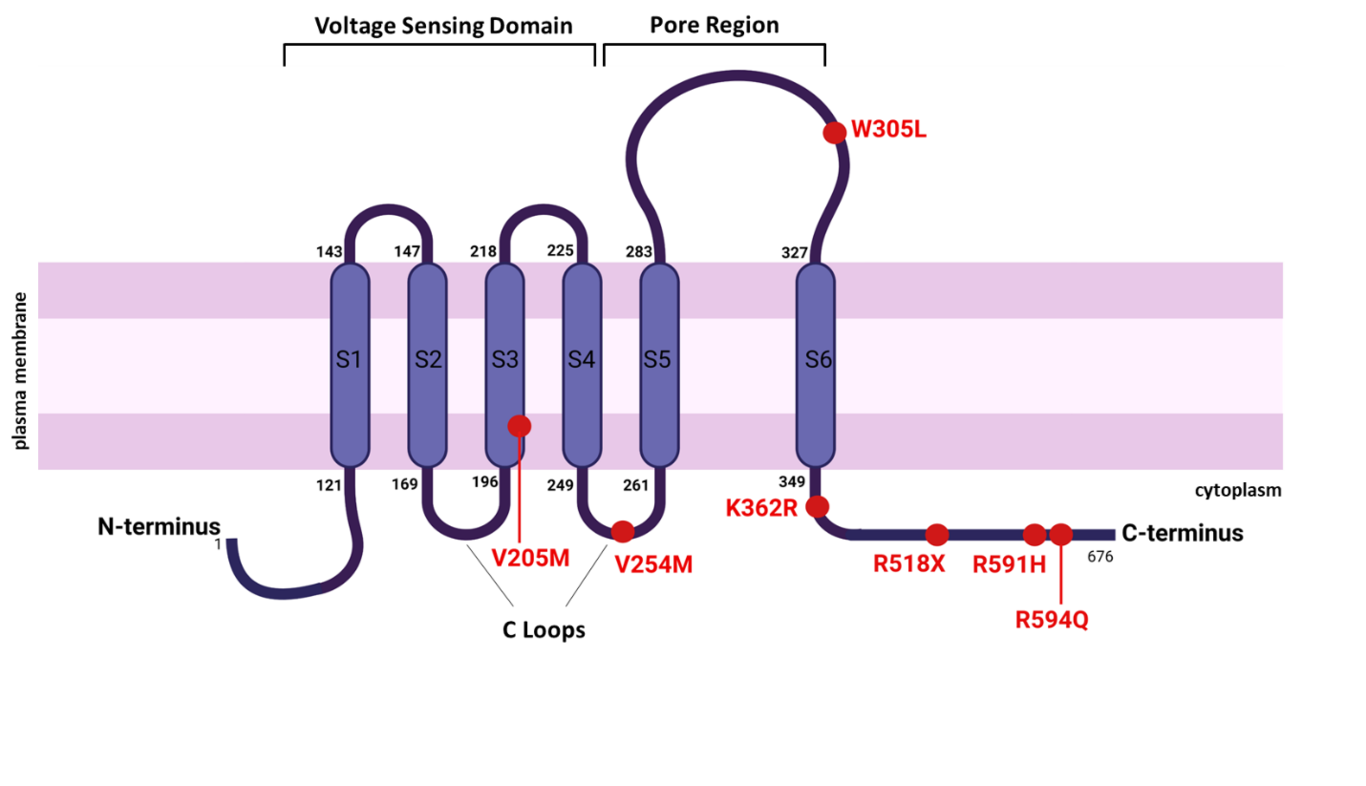
Background: Long QT Syndrome (LQTS) is a rare familial ion channelopathy that may result in syncope, cardiac arrest and sudden death. Ion channel gene variants have been implicated in neurodevelopmental disorders (NDDs), however the link between LQTS and NDDs in children is not well characterized. Methods: This retrospective observational cohort study included patients diagnosed with LQTS at <19 years of age with an NDD diagnosis, prospectively enrolled in an inherited arrhythmia registry at a tertiary hospital between April 2015- June 2021. Patients with hypoxic ischemic injury were excluded. Demographics, genetics, therapy and outcomes were evaluated. Results: Among 106 LQTS patients in the registry, we identified 15 (14%) with NDDs. Eleven (73%) of 15 patients were male compared with 4 (27%) females (p=0.02). Thirteen (87%) were KCNQ1-positive, with mean age at LQTS diagnosis of 6.6 years (SD: 4.3) and baseline QTc of 446ms (SD: 24). Eight (53%) patients had attention deficit hyperactivity disorder, followed by 4 (27%) with learning/communication disorder, 3 (20%) with autism spectrum disorder and 2 (13%) with motor disorder. Nine of 15 (60%) patients received an NDD diagnosis 4.4 (SD: 2.1) years post-LQTS diagnosis; 4 (27%) pre-LQTS diagnosis, and 2 (13%) were unknown. Thirteen (87%) patients were treated with Nadolol monotherapy, 1 (7%) with flecainide and 1 (7%) with lifestyle modifications only. Five (33%) patients were taking a concomitant psychostimulant for their NDD, and none experienced arrhythmic events on therapy. LQTS-related event was experienced by 1 (7%) patient over a mean follow-up of 5.7 (SD: 3.9) years. Conclusion: The prevalence of NDD in LQTS patients (14%) was higher compared to the general population (4.5-9%). Larger studies investigating the link between KCNQ1, other LQTS-related genes and NDDs are warranted.