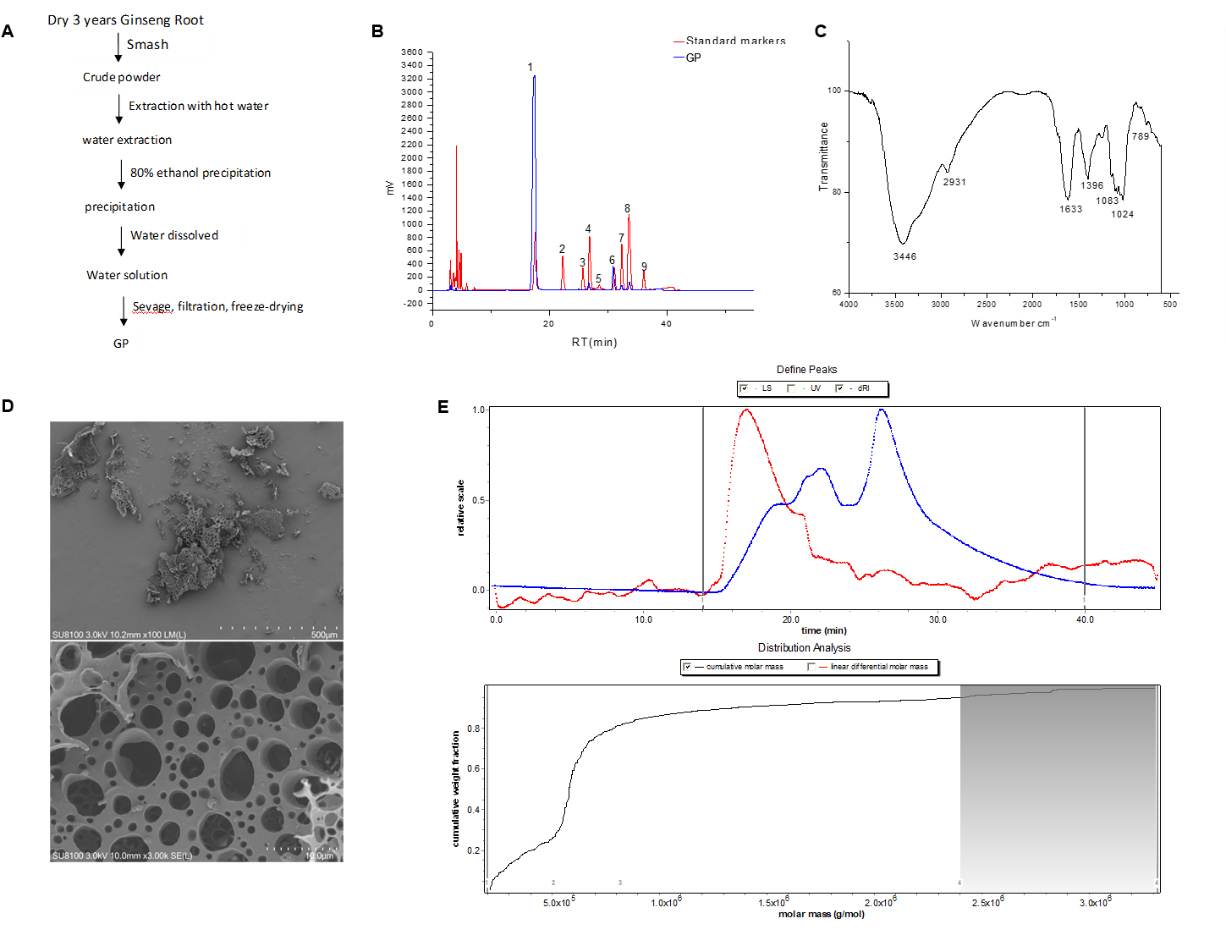
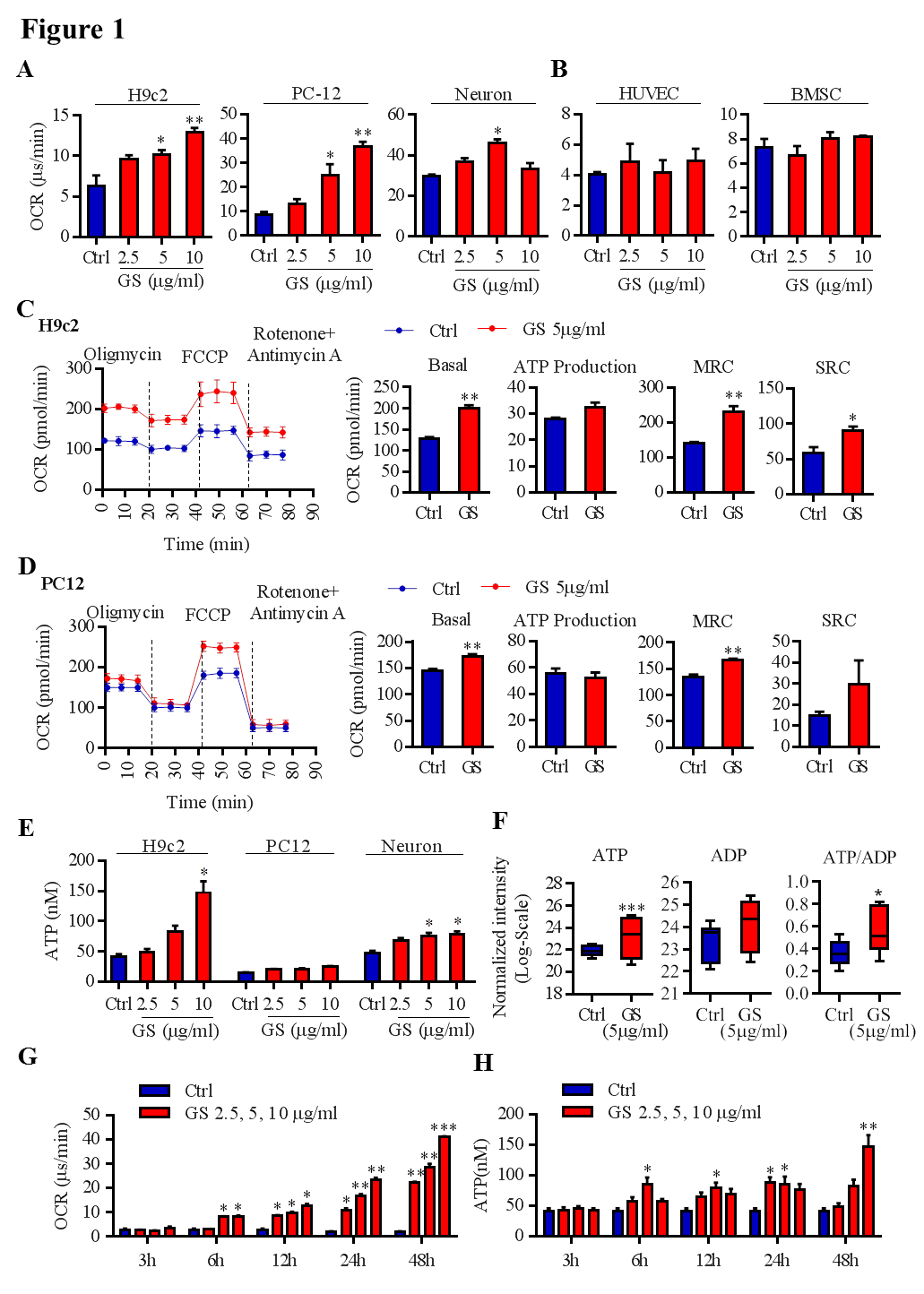
Aerobic cellular respiration provides chemical energy, adenosine triphosphate (ATP), to maintain multiple cellular functions. Sirtuin 1 (SIRT1) can deacetylate peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α) to promote mitochondrial biosynthesis. Targeting energy metabolism is a potential strategy for the prevention and treatment of various diseases, such as cardiac and neurological disorders. Ginsenosides, one of the major bioactive constituents of Panax ginseng, have been extensively used due to their diverse beneficial effects on healthy subjects and patients with different diseases. However, the underlying molecular mechanisms of total ginsenosides (GS) on energy metabolism remain unclear. In this study, oxygen consumption rate, ATP production, mitochondrial biosynthesis, glucose metabolism, and SIRT1-PGC-1α pathways in untreated and GS-treated different cells, fly, and mouse models were investigated. GS pretreatment enhanced mitochondrial respiration capacity and ATP production in aerobic respiration-dominated cardiomyocytes and neurons, and promoted tricarboxylic acid metabolism in cardiomyocytes. Moreover, GS clearly enhanced NAD+-dependent SIRT1 activation to increase mitochondrial biosynthesis in cardiomyocytes and neurons, which was completely abrogated by nicotinamide. In addition, GS had protective effects against hypoxia- or oxygen-glucose deprivation-induced cardiomyocyte damage through activation of the SIRT1-PGC-1α pathway. Importantly, ginsenoside monomers, such as Rg1, Re, Rf, Rb1, Rc, Rh1, Rb2, and Rb3, were found to activate SIRT1 and promote energy metabolism. This study may provide new insights into the extensive application of ginseng for cardiac and neurological protection in healthy subjects and patients with ischemic disorders.