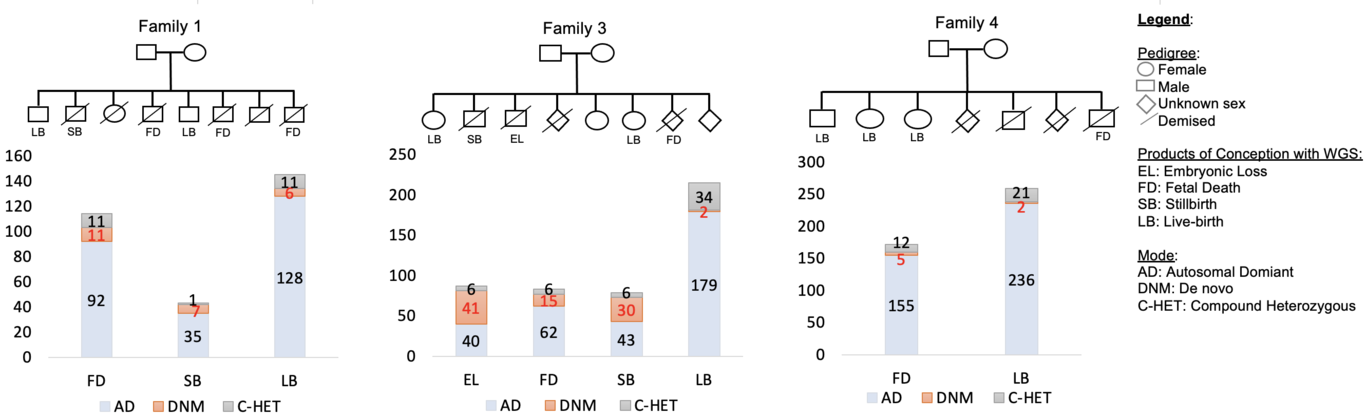
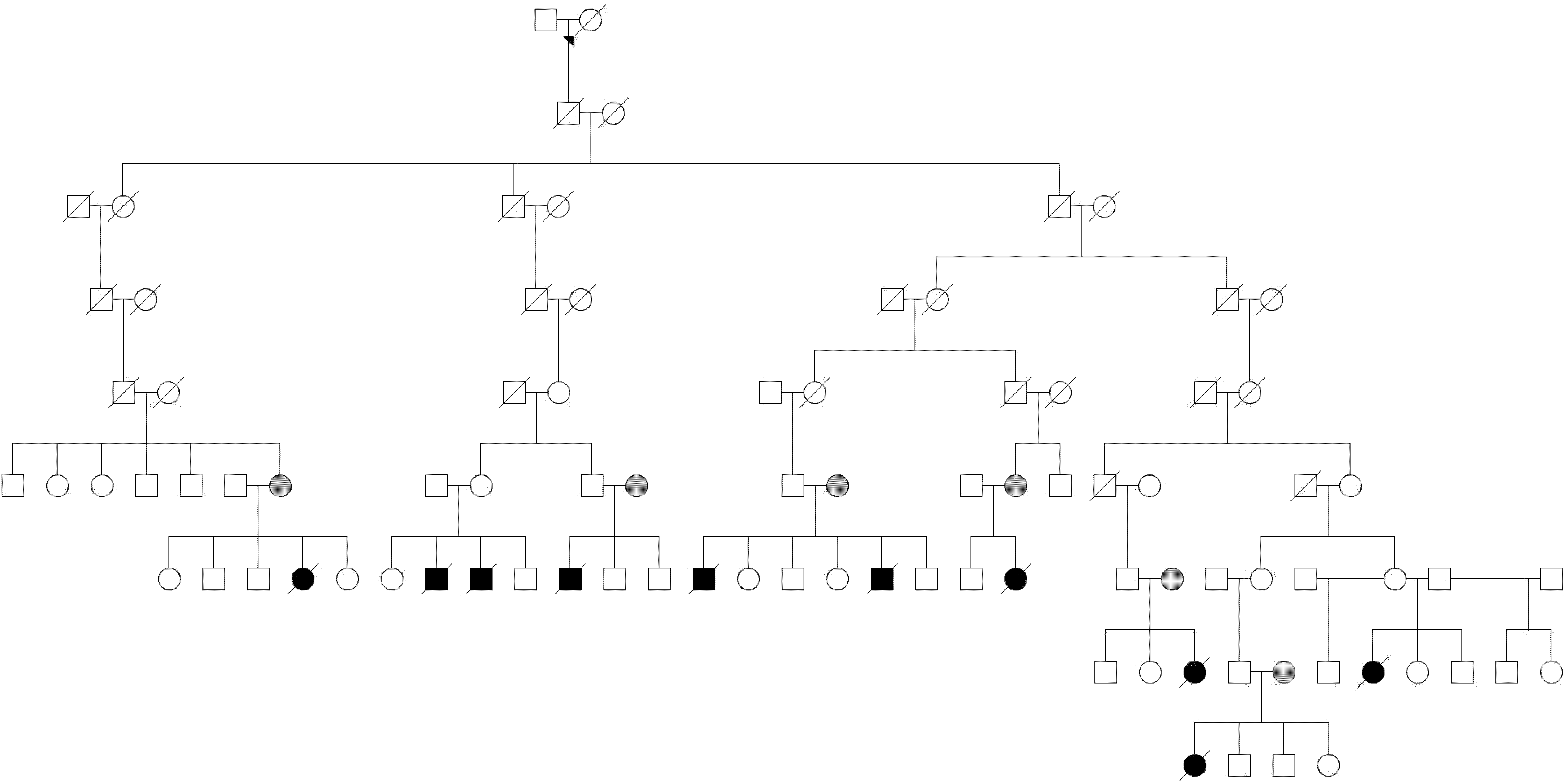Objective To conduct a feasibility whole-genome sequencing (WGS) study in families to identify genetic variants relevant to unexplained pregnancy loss. Methods We conducted a pilot WGS study of four families with recurrent pregnancy loss, including parents, healthy live births, and losses, which included an embryonic loss (<10 weeks’ gestation), fetal deaths (10-20 weeks’ gestation) and stillbirths (≥ 20 weeks’ gestation). We used the Illumina platform for WGS and state-of-the-art protocols to identify single nucleotide variants (SNVs) following various modes of inheritance. Results We identified 87 SNVs involving 75 genes in embryonic loss (n=1), 370 SNVs involving 228 genes in fetal death (n=3), and 122 SNVs involving 122 genes in stillbirth (n=2). Of these, 22 de novo, 6 autosomal dominant and an X-linked recessive SNVs were pathogenic (probability of being loss-of-function intolerant >0.9), impacting known genes (e.g., DICER1, FBN2, FLT4, HERC1, and TAOK1) involved in embryonic/fetal development and congenital abnormalities. Further, we identified missense compound heterozygous SNVs impacting genes (e.g., VWA5B2) in two fetal death samples that were absent from live births and population controls, providing evidence for haplosufficient genes relevant to pregnancy loss. Conclusions In this pilot study, we provide evidence for de novo and inherited SNVs relevant to pregnancy loss. Our findings provide justification for conducting WGS using larger numbers of families and warrant validation by targeted sequencing to ascertain causal variants. Elucidating genes causing pregnancy loss may facilitate the development of risk stratification strategies and novel therapeutics.