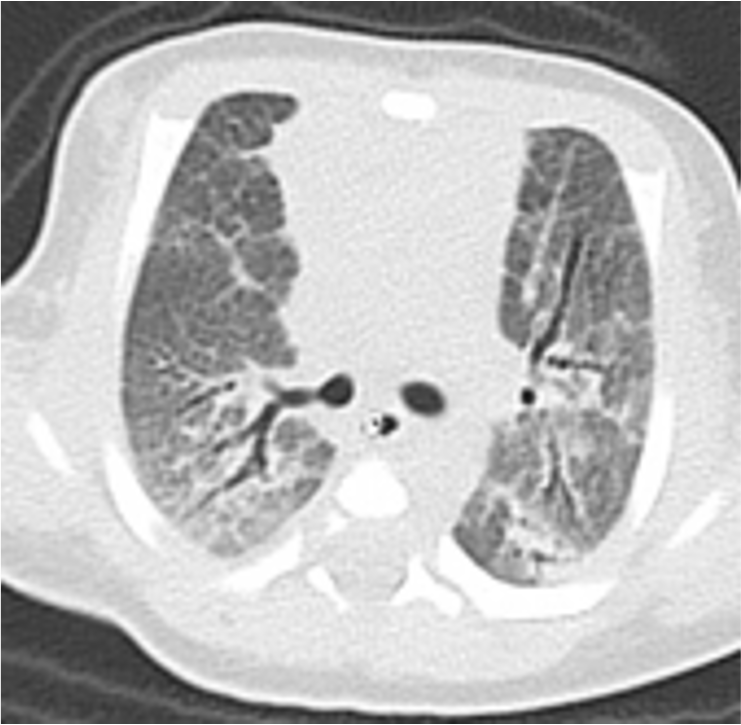
The authors declare no conflicts of interest.Financial Support: Japan Society for the Promotion of Science 19K08299Keywords: Patients with Hermansky–Pudlak syndrome, Pulmonary fibrosis,AP3B1 mutation, Hemophagocytic lymphohistiocytosis, NeonatalTo the Editor,Hermansky–Pudlak syndrome (HPS) is an inherited disorder characterized by albinism of the oculocutaneous region and hemorrhagic disease. Eleven genes causative of HPS have been identified, and the clinical phenotypes of the condition differ depending on which gene is affected. HPS type 2 (HPS2) is an autosomal recessive inherited disease caused by mutations in AP3B1 , resulting in pulmonary fibrosis (PF) and immunodeficiency1. Patients with HPS2 also have neutropenia and decreased NK cell cytotoxicity, which can lead to hemophagocytic lymphohistiocytosis (HLH); however, the total number of HPS2 cases reported was less than 40 as of March 20211, making it difficult to accurately assess the risk of HLH associated with this disease. In addition, although PF occurs earlier in HPS2 than in other types of HPS, HPS2 with PF in the neonatal period has never been reported. Here, we report a case of HPS2 with neonatal PF harboring mutations in both AP3B1 alleles, one of which is novel.The patient is female, the second child of a non-consanguineous marriage, and was born at 39 weeks 4 days (birth weight, 3798 g) by spontaneous vaginal delivery. Apgar score was 9 at 1 minute; however, respiratory failure gradually developed, resulting in endotracheal intubation and mechanical ventilation. At 20 hours after birth, a right tension pneumothorax developed, which required thoracentesis, thoracic drainage, and nitric oxide inhalation therapy. A computerized tomography scan at 6 days of age revealed PF (Fig. 1). Septic workup, including blood and throat swabs, was negative. Respiratory distress syndrome was less likely due to term delivery; therefore, we suspected hereditary interstitial lung disease and tested the SFTPB , SFTPC ,ABCA3 , FOXF1 , NKX2.1 , GATA2 , CSF2RA , and CSF2RB genes, all of which were negative2. Since she had albinism, we performed genetic analysis of 22 albinism-related genes, which revealed two heterozygous variants inAP3B1 : c.1122_1123insAG (p.Phe375fs*) and c.2546T>A (p.Leu849Ter). The former variant is a novel frameshift mutation, and the latter is a nonsense mutation that was recently reported as the causative mutation in a patient with HPS2 in Japan3. Neither variant is recorded in the Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/) database, and both are considered pathological. Each parent carried one of the variants; therefore, we diagnosed the patient with HPS2 caused by AP3B1 mutations. The patient also suffered from mitral regurgitation, which did not require additional medication.The patient’s respiratory condition improved, and she was discharged from hospital at 190 days of age, with the introduction of home oxygen therapy. At 9 months old, she was hospitalized due to HLH. Physical examination revealed no abnormal findings, other than pharyngeal injection and slight fever. Laboratory tests showed thrombocytopenia, with platelets 3.0 × 104/μL and white blood cell count 1100/μL (2.8% neutrophils). Ferritin was not raised; however, soluble IL2 receptor levels were elevated at 7469 U/mL. Bone marrow examination showed evidence of hemophagocytosis. CD107a expression was decreased in CD3–CD56+ NK cells compared with control NK cells, detected as previously described4, suggesting a decrease in cytotoxic degranulation (Fig. 2). Blood samples were negative for Epstein–Barr virus, cytomegalovirus, and human parvovirus B12. The patient was diagnosed with HLH associated with viral infection. A primary immunodeficiency gene panel, comprising approximately 400 genes, was evaluated; no mutations in PFR1 ,UNC13D , STX11 , STXBP2 , RAB27A , orLYST were detected. She was treated with dexamethasone (10 mg/m2/day) and immunoglobulin (2.5 g/dose), with good clinical response. We started G-CSF for continuous neutropenia after recovery from HLH. She had another episode of fever with thrombocytopenia due to viral infections at the age of 1 year 9 months.Among the primary immunodeficiency diseases affecting cytotoxic granules, patients with hereditary granulopathies, such as Chediak–Higashi and Griscelli syndrome type 2, are at high risk of severe HLH, and early hemopoietic stem cell transplantation (HSCT) is a treatment option5. By contrast, complication of HLH occurs less frequently in HPS2, although there is a risk of developing HLH in some patients with the condition. Jessen et al. reported that the risk of developing HLH was relatively low in a mouse HPS2 model affecting Ap3b1 (the pearl mouse)6, which is compatible with reports regarding patients with HPS2; however, Dell’Acqua et al. cautioned of the risk of HLH in HPS2, in their report of a case with lethal HLH7.Further delineation of genotype–phenotype correlations betweenAP3B1 mutations and complication of HLH indicated that truncating mutations, rather than non-truncating changes such as missense mutations, may be associated with a higher risk of developing HLH, which is consistent with our case having nonsense and frameshift mutations inAP3B1. Regarding genotype–phenotype correlation of PF in HPS2, 17 of 37 cases are reported to have developed PF, all of which had homozygous or compound heterozygous truncating mutations. The lack of missense mutations among HPS2 cases with HLH and/or PF suggests that missense variants may be less pathogenic, in terms of HLH and PF. It will be necessary to accumulate additional cases of HPS2 and follow their clinical courses in detail to understand these genotype–phenotype correlations more precisely.To the best of our knowledge, this is the first case of HPS2 with PF observed in the neonatal period. Among types of HPS, PF occurs mainly in HPS1, HPS2, and HPS4. In HPS1 (the most frequent type), 100% of patients develop PF in their thirties or forties8. Hengst et al. reported six cases of HPS2 with PF, with a mean age of 8.8 years, respiratory symptoms 3.3 years before diagnosis, and a minimum age of 0.8 years9. It has not previously been reported that patients with HPS2 suffer from mitral regurgitation, which may contribute to early onset of PF. In any case, the onset of PF in our case is the earliest recorded to date, and suggests that neonatologists should consider HPS2 as a diagnosis for neonates presenting with PF and albinism.In conclusion, we report a case of HPS2 with neonatal-onset PF and an episode of HLH, caused by truncating compound heterozygous mutations of AP3B1 . Clinicians should be aware of the importance of accurate genetic analysis of AP3B1 to evaluate the severity of clinical phenotypes, particularly the onset of PF and morbidity from HLH.Keigo Matusyuki1Mizuki Ide1Keishirou Houjou1Saho Shima1Seiji Tanaka1Yoriko Watanabe1Tomoko Egashira2Hiroyuki Tomino2Toshimitsu Takayanagi2Tashiro Katsuya3Ken Okamura4Tamio Suzuki4Ryuta Nishikomori11Department of Pediatrics and Child Health, Kurume University School of MedicineKurume, Fukuoka, Japan2National Hospital Organization Saga National HospitalSaga, Saga, Japan3Karatsu Red Cross HospitalKaratu, Saga, Japan4Department of Dermatology, Faculty of Medicine, Yamagata UniversityYamagata, Yamagata, JapanAcknowledgmentsWe thank Ms. Ohhinata for technical assistance.Key Message: Hermansky–Pudlak Syndrome type2 can develop pulmonary fibrosis in the neonatal periodReferences1. Huizing M, Malicdan M, Gochuico B, Gahl W. Hermansky-Pudlak Syndrome.GeneReviews 2000; https://www.ncbi.nlm.nih.gov/books/NBK1287/, may 2021.2. Okamura K, Hayashi M, Abe Y, et al. NGS-based targeted resequencing identified rare subtypes of albinism: Providing accurate molecular diagnosis for Japanese patients with albinism. Pigment Cell Melanoma Res. 2019;32(6):848-853.3. Nishikawa T, Okamura K, Moriyama M, et al. Novel AP3B1 compound heterozygous mutations in a Japanese patient with Hermansky-Pudlak syndrome type 2. J Dermatol. 2020;47(2):185-189.4. Shibata H, Yasumi T, Shimodera S, et al. Human CTL-based functional analysis shows the reliability of a munc13-4 protein expression assay for FHL3 diagnosis. Blood. 2018;131(18):2016-2025.5. Sharma P, Nicoli ER, Serra-Vinardell J, et al. Chediak-Higashi syndrome: a review of the past, present, and future. Drug Discov Today Dis Models. 2020;31:31-36.6. Jessen B, Bode SF, Ammann S, et al. The risk of hemophagocytic lymphohistiocytosis in Hermansky-Pudlak syndrome type 2. Blood.2013;121(15):2943-2951.7. Dell’Acqua F, Saettini F, Castelli I, Badolato R, Notarangelo LD, Rizzari C. Hermansky-Pudlak syndrome type II and lethal hemophagocytic lymphohistiocytosis: Case description and review of the literature.J Allergy Clin Immunol Pract. 2019;7(7):2476-2478 e2475.8. Vicary GW, Vergne Y, Santiago-Cornier A, Young LR, Roman J. Pulmonary Fibrosis in Hermansky-Pudlak Syndrome. Ann Am Thorac Soc.2016;13(10):1839-1846.9. Hengst M, Naehrlich L, Mahavadi P, et al. Hermansky-Pudlak syndrome type 2 manifests with fibrosing lung disease early in childhood.Orphanet J Rare Dis. 2018;13(1):42.Figure legendsFig 1. CT image of interstitial pneumonia at 6 days of age.Bilateral lung fields showing diffuse ground-glass opacity and bronchioles dilated to obliteration. Some dorsal lung fields are hyperintense, possibly reflecting the effect of gravity.Fig 2. Patient genotype and NK cell activity.(a) Chromatogram demonstrating compound heterozygous mutations, c.1122_1123insAG (p.Phe375fs)* and c.2546T>A (p.Leu849Ter), in AP3B1 .(b) Analysis of NK cell degranulation by flow cytometric analysis. Profiles show CD56 versus CD107a staining on CD3−CD56+NK cells after stimulation. CD107a expression was significantly decreased in patient cells (above) relative to control cells (below). The assay was performed as previously described4.