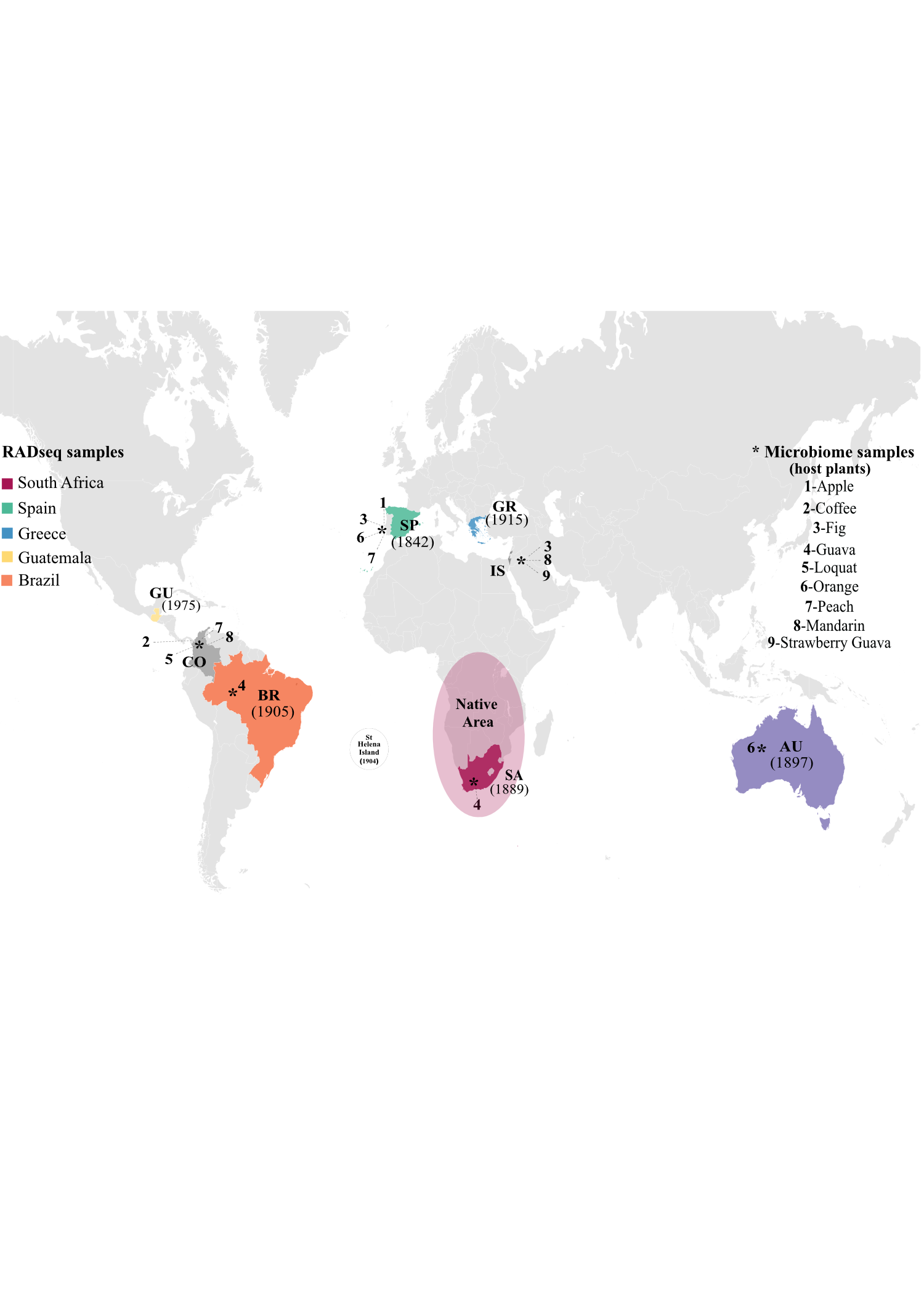
Invasive species are among the most important, growing threats to food security and agricultural systems. The Mediterranean fruit fly Ceratitis capitata is one of the most damaging representatives of a group of rapidly expanding species in the family Tephritidae due to their wide host range and high invasiveness. Here, we used restriction site-associated DNA sequencing (RADseq) to investigate population genomic structure and phylogeographic history of medflies collected from six sampling sites, including Africa (South Africa), the Mediterranean (Spain, Greece), Latin America (Guatemala, Brazil) and Australia. A total of 1,907 single nucleotide polymorphisms (SNPs) showed two genetic clusters separating native and introduced ranges, consistent with previous findings. In the introduced range, all individuals were assigned to one genetic cluster except for those in Brazil, which showed introgression of a genetic cluster that also appeared exclusively in South Africa and could not be previously identified using microsatellite markers. Moreover, the microbiome variations in medfly populations from selected sampling sites was assessed by amplicon sequencing of the 16S ribosomal RNA (V4 region). No strong patterns of microbiome variation were detected across geographic regions or host plants, except for the differentiation of the Brazilian specimens which showed increased diversity and unique composition of its microbiome compared to other sampling sites. The unique SNP patterns in the Brazilian specimens could point to a direct migration route from Africa with subsequent adaptation of the microbiota to the specific conditions present in Brazil. These findings significantly improve our understanding of the evolutionary history of global medfly invasions and adaptation to newly colonised environments.