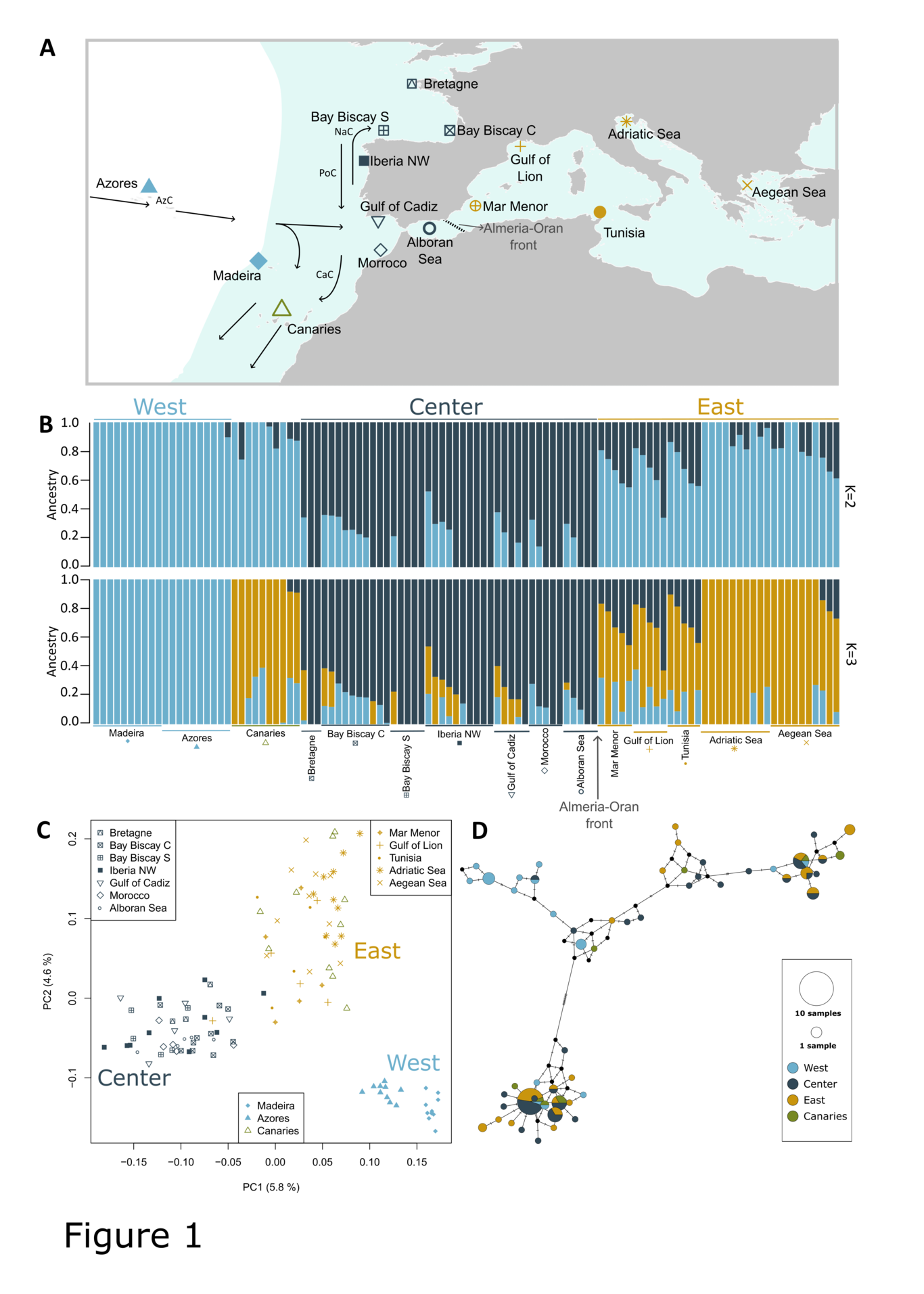
Next-generation sequencing of pooled samples (Pool-seq) is a popular method to assess genome-wide diversity patterns in natural and experimental populations. However, Pool-seq is associated with specific sources of noise, such as unequal individual contributions. Consequently, using Pool-seq for the reconstruction of evolutionary history has remained underexplored. Here we describe a novel Approximate Bayesian Computation (ABC) method to infer demographic history, explicitly modeling Pool-seq sources of error. By jointly modeling Pool-seq data, demographic history and the effects of selection due to barrier loci, we obtain estimates of demographic history parameters accounting for technical errors associated with Pool-seq. Our ABC approach is computationally efficient as it relies on simulating subsets of loci (rather than the whole-genome), and on using relative summary statistics and relative model parameters. Our simulation study results indicate Pool-seq data allows distinction between general scenarios of ecotype formation (single versus parallel origin), and to infer relevant demographic parameters (e.g., effective sizes, split times). We exemplify the application of our method to Pool-seq data from the rocky-shore gastropod Littorina saxatilis, sampled on a narrow geographical scale at two Swedish locations where two ecotypes (Wave and Crab) are found. Our model choice and parameter estimates show that ecotypes formed before colonization of the two locations (i.e., single origin) and are maintained despite gene flow. These results indicate that demographic modeling and inference can be successful based on pool-sequencing using ABC, contributing to the development of suitable null models that allow for a better understanding of the genetic basis of divergent adaptation.