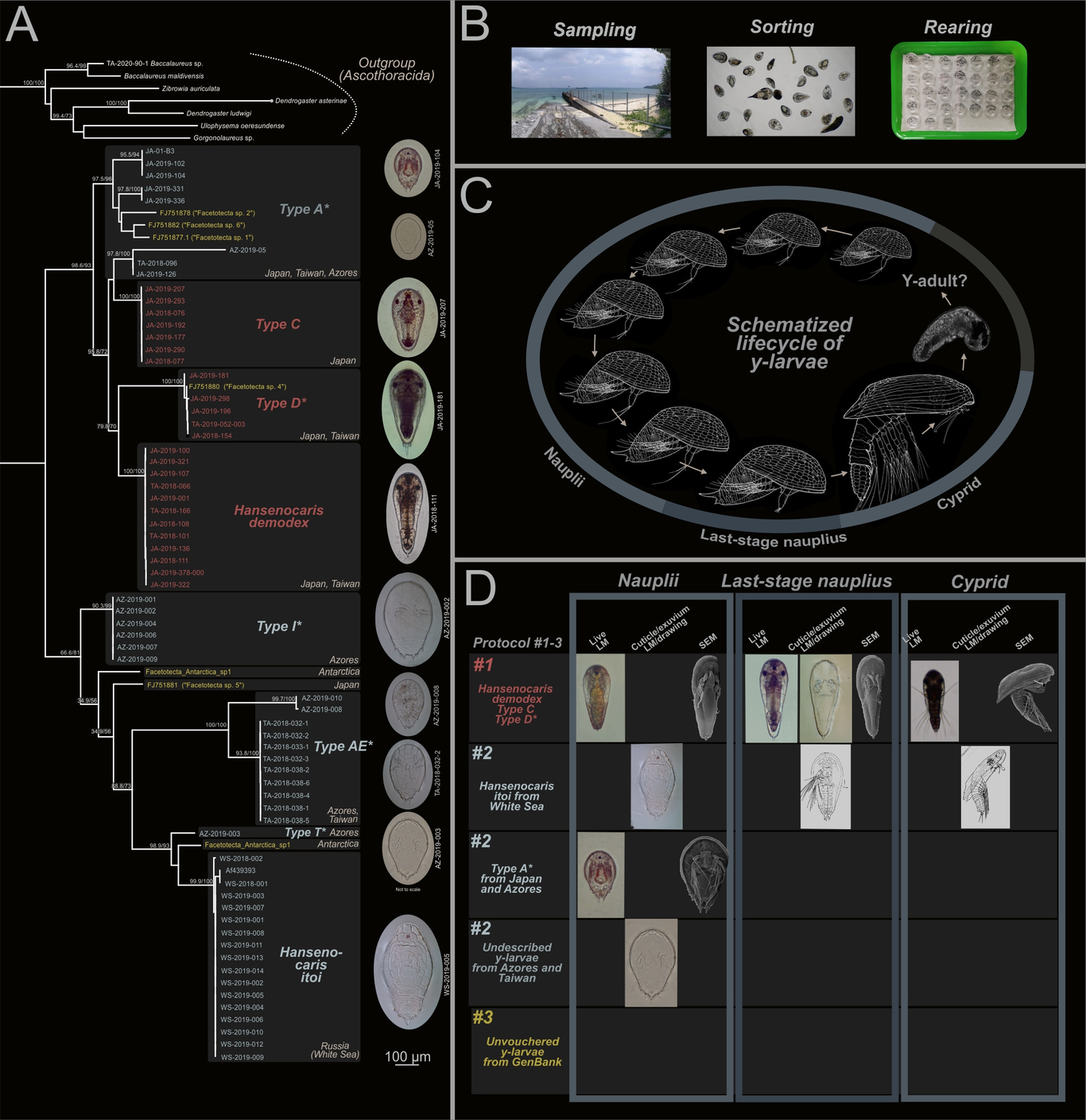
The enigmatic “y-larvae” (Pancrustacea: Facetotecta) still have an incompletely understood lifecycle, and their adult forms remain unknown despite their discovery more than 100 years ago and their documented global occurrence from shallow waters to the deep-sea. Only two of the 17 formally described species, all based on larval stages, have been investigated using an integrative taxonomic approach that, besides providing descriptions of the morphology of the naupliar and cyprid stages, also made use of exuvial voucher material and DNA barcodes. To improve our knowledge about the systematics and phylogenetics of y-larvae, we developed a novel protocol that maximizes the amount of morphological, ecological, and molecular data that can be harvested from single individuals of these tiny larvae. This revolves around single larva barcoding, and includes daily imaging of y-nauplii reared in culture dishes, mounting of their last naupliar exuviae on a slide as a reference voucher, live imaging of the y-cyprid instar that follows, and fixation, DNA extraction, amplification, and sequencing of the y-cyprid specimen. By developing and testing a suite of new primers for both nuclear and mitochondrial protein-coding and ribosomal genes, we estimated the most comprehensive phylogeny of Facetotecta to date. We expect that our novel procedure will help to unravel the complex systematics of y-larvae and show how these fascinating larval forms have evolved. Moreover, we posit that our protocols should work on larval specimens of a diverse array of molting marine invertebrate taxa.