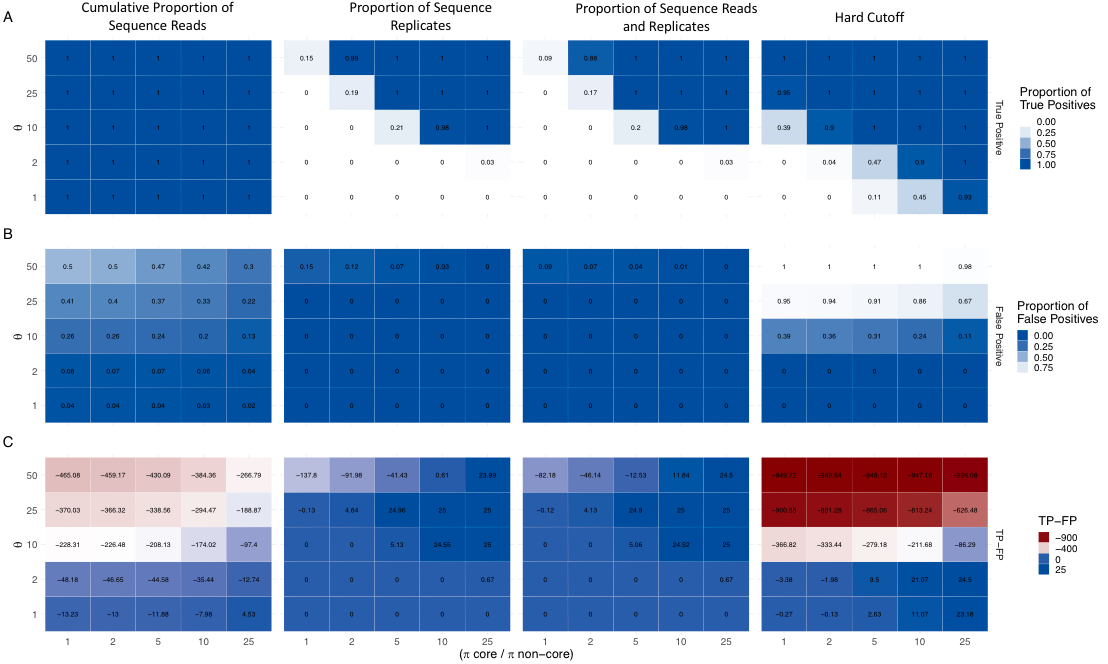
Community ecology includes linking variation in system functions to the distribution and abundance of taxa. In inferring processes, functions, and causal taxa, it is common practice to assume a core community can be defined and that attributes of the core are representative of the entire dataset. Assuming categorical thresholds in abundance exist has the potential to be misleading, especially if rare taxa are contributing to ecological processes. Additionally, there are no standard criteria for core membership, complicating comparisons across studies. Rather, the existence of a core set of taxa can be treated as a hypothesis that may or may not be supported. We considered four methods commonly used for defining a core in studies of microbiomes and applied them to two published microbial data sets and simulations covering a range of plausible communities. We evaluated the ability of each method to correctly categorize taxa. Assignment of core taxa varied substantially among methods and datasets. Additionally, the ability of evaluated methods to capture the simulated core was contingent on the distribution of taxon abundances. While able to correctly identify core taxa in select cases, the methods disagreed more often than not. Given the lack of agreement among core assignment methods, categorization of taxa into sets corresponding to core and non-core is questionable and requires testing and validation before use in any particular context. Our results do not support applying methods of dimension reduction for core taxa classification, but instead provide additional rationale to favor analyses that use abundance data in their entirety.