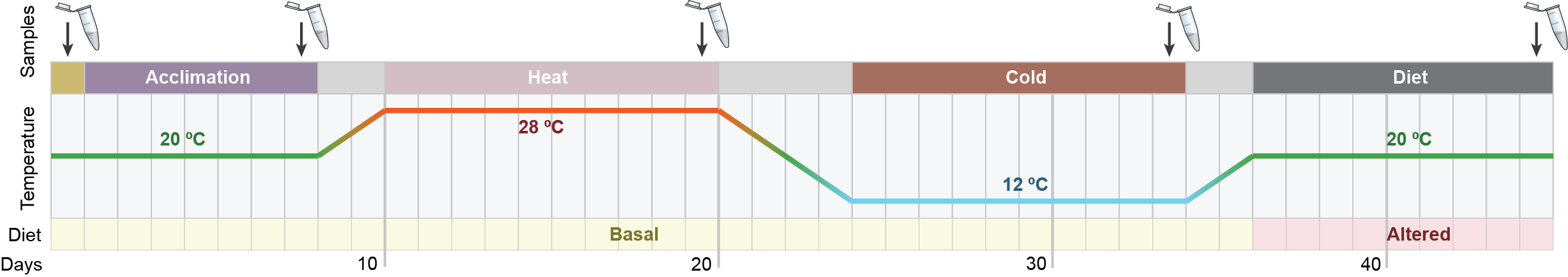
The gut microbiomes that associate with animals can represent labile units of cooperating and competing microbes. This lability, sometimes referred to as metagenomic plasticity, has been posited to have an important role as an additional axis of hosts’ phenotypic plasticity. However, whether and how metagenomic plasticity varies across hosts with different ecological and evolutionary features remains unclear. To address this, we utilised faecal-derived genome-resolved metagenomics and compared how the taxonomic, phylogenetic and functional microbial dynamics varied across a series of disturbances in two mammal species; namely, the insectivorous-specialist, Crocidura russula (N = 29) and the omnivorous-generalist Apodemus sylvaticus (N = 22). Although faecal microbial diversity of both species remained stable, compositional dynamics differed significantly. C. russula exhibited substantially higher variability and directionality of microbial responses, with higher predictability associated with each disturbance, compared to A. sylvaticus. Predictions of functional traits using joint-species distribution modelling supported these observations. C. russula showed strong functional response to perturbations, with marked directional variation of various metabolic functions. In contrast, the significantly higher functional diversity and redundancy of the A. sylvaticus microbiome likely buffered its functional response to perturbations, which remained more constant across time. Our results indicate that the intrinsic properties (e.g., diversity, redundancy) of gut microbiomes associated with animals with different biological attributes shape the taxonomic, phylogenetic, and functional response to environmental stressors. This level of plasticity might affect the capacity of animal hosts to acclimate and adapt to changing environments.