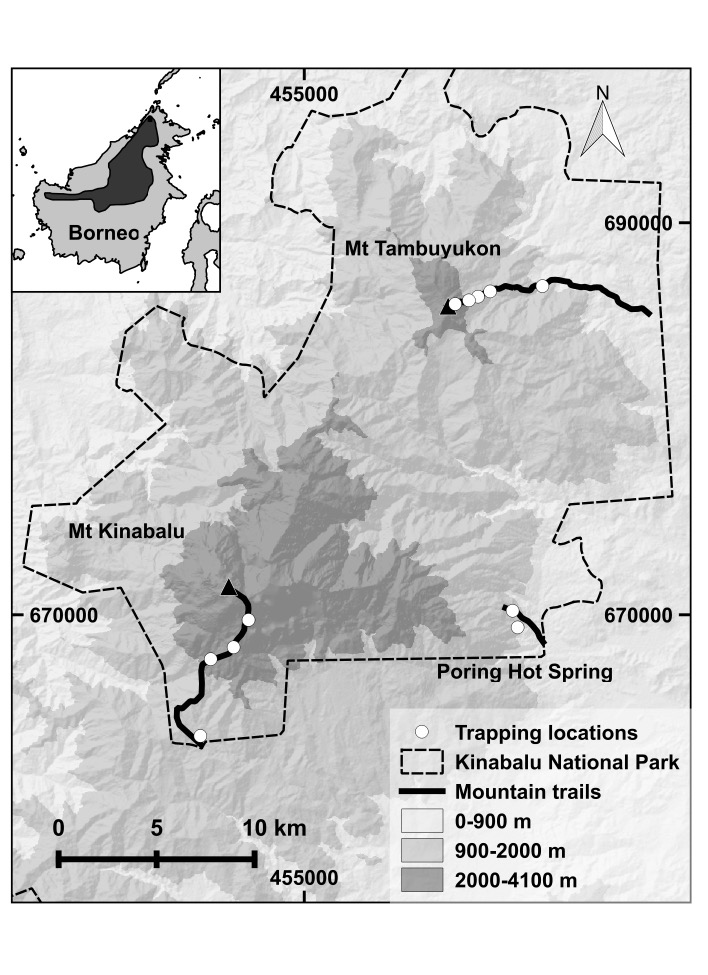
The use of museum specimens held in natural history repositories for population and conservation genetic research is increasing in tandem with the use of next generation sequencing technologies. Short Tandem Repeats (STRs), or microsatellite loci, are commonly used genetic markers in population and conservation genetic studies. However, they traditionally suffered from a host of issues: fragment size homology, high costs, and low throughput as a result of capillary electrophoresis genotyping and difficulty in reproducibility across laboratories. Next generation sequencing technologies can address these problems, but the incorporation of DNA derived from museum specimens suffers from significant fragmentation and contamination with exogenous DNA. Combatting these issues requires extra measures of stringency in the lab and during data analysis, yet there have not been any studies evaluating microsatellite allelic dropout from museum specimen extracted DNA. In this study, we explore a high throughput sequencing method to evaluate the amount of variation found within museum specimen DNA extracts for previously characterized microsatellites across PCR replicates. We found it useful to classify samples based on quality after replicated PCRs, which determined the rate by which genotypes were accurately recovered. We also found that longer microsatellites performed worse in all museum specimens, so when designing a study invoking museum specimens, short markers (under 250 bp) should be preferentially selected. Allelic dropout rates across loci were dependent on sample quality. The high quality museum specimens performed as well, and recovered nearly as high quality metrics as our tissue sample. Mitochondrial DNA sequences were not predictive of nuclear DNA presence, as all samples recovered cytochrome b fragments yet many lacked microsatellite genotypes, particularly in samples deemed low quality. Based on our results, we have provided a set of best practices for screening, quality assurance, and incorporation of reliable genotypes from museum specimens.