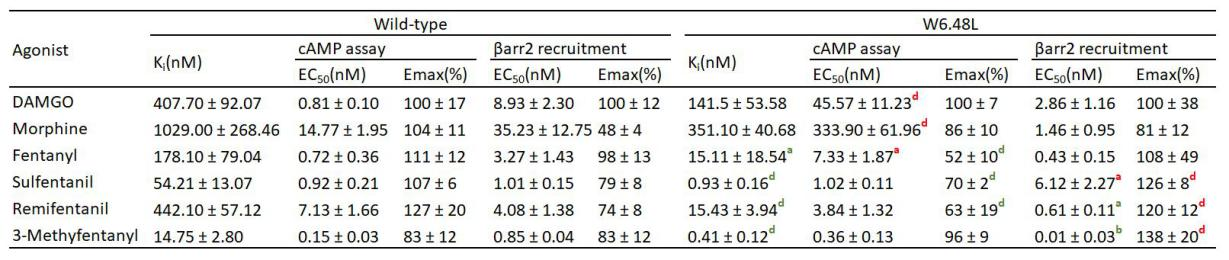
Background and Purpose: β-arrestin2 plays an important role in opioid receptor signaling, but its involvement in morphine- and fentanyl-induced respiratory depression is widely debated. The aim of this study was to determine whether β-arrestin2 signaling is associated with fentanyl- and morphine-induced respiratory depression. Experimental Approach: This study investigated whether β-arrestin2 is involved in respiratory depression induced by fentanyl or morphine by inhibiting the upstream signaling molecule GRK2 and knocking out β-arrestin2 in mice, using whole-body plethysmography chambers to assess changes in respiratory function. Key Results: In the experiment of inhibiting GRK molecules, GRK inhibitors significantly improved the respiratory depression induced by morphine, but had no effect on fentanyl. In experiment of knocking out β-arrestin2, respiratory depression was significantly improved in the morphine group, but less affected in the fentanyl group. Conclusion and Implications: Our results suggested that inhibition of β-arrestin2 signaling alleviated morphine-induced respiratory depression but had little effect on fentanyl-induced respiratory depression in both models, suggesting differences in the respiratory depression mechanisms between fentanyl and morphine. This suggests that we may need to give a differentiated dosing regimen in clinical treatment of respiratory depression caused by the two drugs.