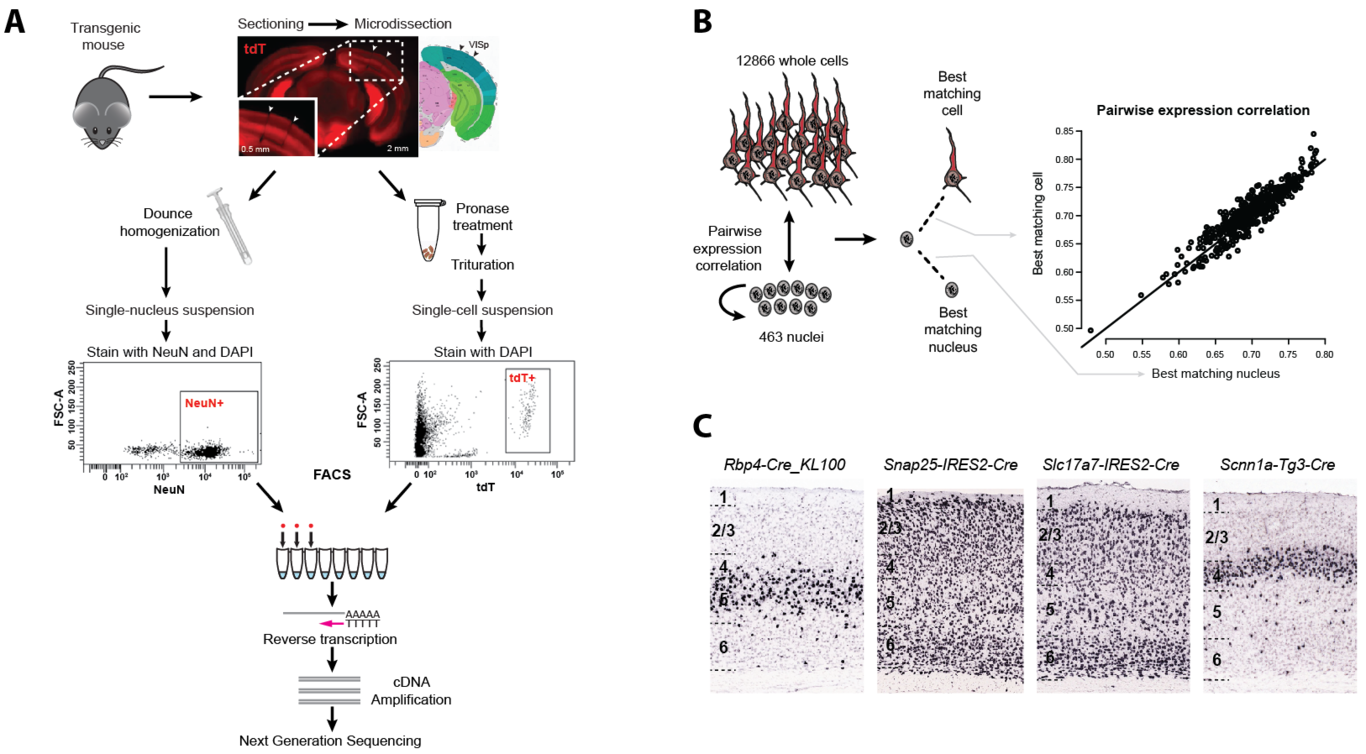
Transcriptomic profiling of complex tissues by single-nucleus RNA-sequencing (snRNA-seq) presents some advantages over single-cell RNA-sequencing (scRNA-seq). snRNA-seq provides less biased cellular coverage, does not appear to suffer cell isolation-based transcriptional artifacts, and can be applied to archived frozen specimens. We used well-matched snRNA-seq and scRNA-seq datasets from mouse visual cortex to demonstrate that similarly high cell type resolution of closely related neuronal types can be achieved with both methods if intronic sequences are included in snRNA-seq analysis. More transcripts are detected in individual whole cells (~11,000 genes) than nuclei (~7,000 genes), but the majority of genes have similar detection across cells and nuclei. We estimate that the nuclear proportion of total cellular mRNA varies from 20% to over 50% for large and small pyramidal neurons, respectively. Together, these results illustrate the high information content of nuclear RNA for characterization of cellular diversity in brain tissues.