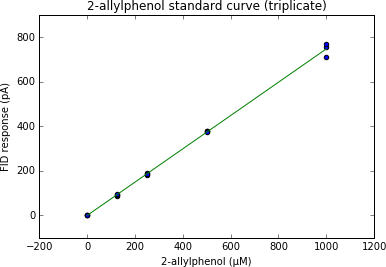
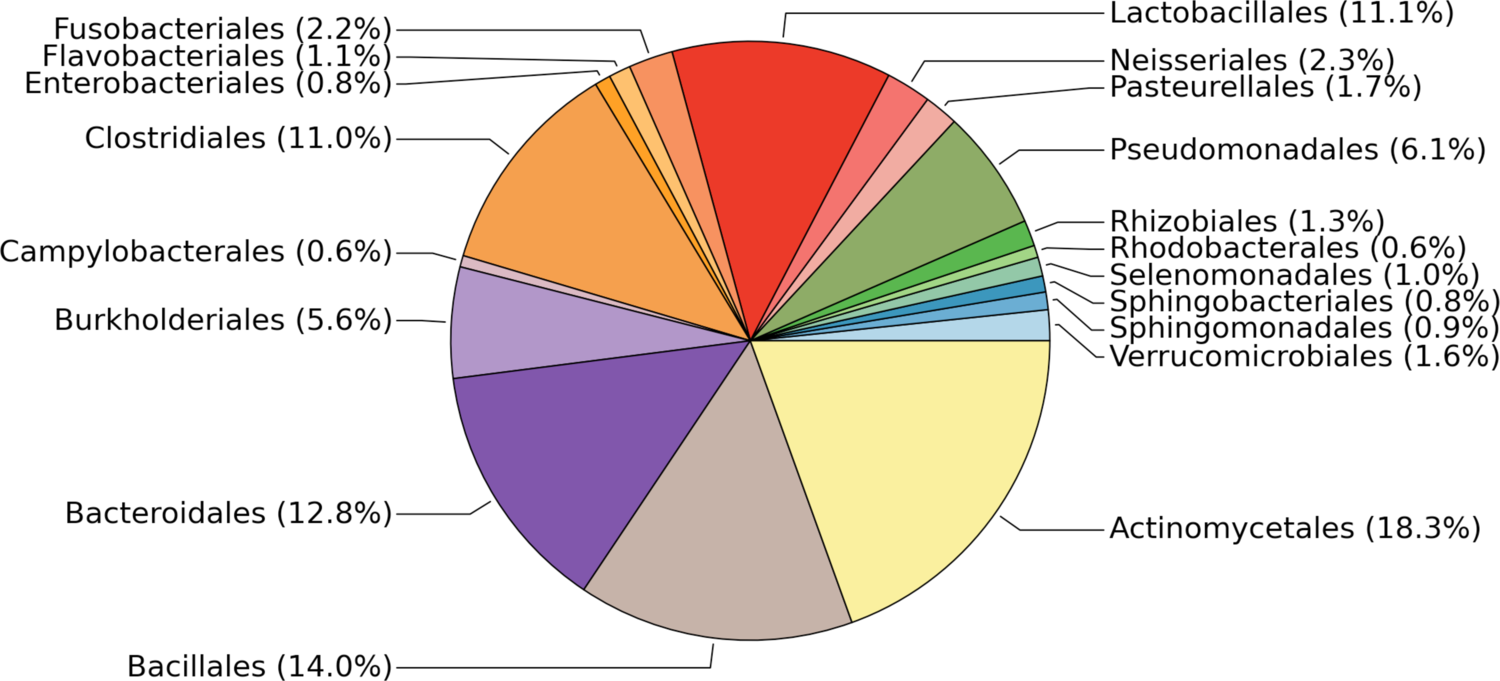
BackgroundGiven an organism to study with RNA-seq, it may have a reference genome or not. In either case, why can't we annotate everything else to the quality of the human or mouse annotations?Considerations in this review will include software, pipelines, problems and best practices.WorkflowThe overall workflow of data->results will serve as the organization for the paper. The focus is not about each step of this workflow (see Conesa et al. 2016). Instead, how will each step affect the annotation?1. Data acquisition: Library prep:Effect of different library preps – both (comment 6 in discussion) - poly(A)-selection - ribo-zero (ribosomal subtraction)Type of RNA - Coding - Non_coding: Non-coding RNA (comment 9 in discussion)RNA-seq Data type: - ONP - PacBio - IlluminaCost benefits of PacBio / Oxford Nanopore sequencing (24e, 2. Pre-processing :Filtration of RNAseq transcriptomes – both (7,- quality trimming- adapter trimmingBest practices (Matt McManus' paper): Less trimming, the better- diginorm: helps with low-coverage discovery vs. reference-based will cause to be more fragmented, and sometimes lose junctions between exons (unpublished horse transcriptome)3. Assembly, split the paper into 2 categories: - reference-based: quality of genome is limiting factor, concept needs to be developed that says if your genome quality is good, then you should do reference-based mapping, or if your quality is poor, then do de novo assembly Effect of genome quality on transcriptome assembly – both (4,5,24a,27Review of Genome-based annotation pipelines (3,15, - pipelines: e.g. Maker & PASA - de novo assembly4. Annotation: this is the meat of what we're talking about in the paper: How to give your gene a name (12, 13,16,18,21,22,23It's a mess. - spotlight the mess - plan for how to solving the messSoftware: dammit, Trinotate5. Databases and Archiving- No universal formatting for description column in gtf, affects downstream analysesMajor genome annotation databases (2,19,26, 24c,29Functional annotations (8,11,14,20,24b,28, 24f, 24gAssignments for everyone:Write down examples faced with trouble with annotation:- getting (archiving)- applying- using downstreamIf you were able to solve, what is the best way to go around?Coordinate in groups:reference-based:- Daniel- Erica- Husseinde novo assembly:- Lisa- Harriet- Tessa- Camille