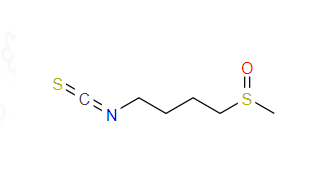
Sulforaphane is the plant active substance with the best anticancer effect and antioxidant found in vegetables. In this paper, the DFT-B3LYP/cc-pVTZ theoretical level was used to optimize the molecular structure of sulforaphane in gas phase, the DLPNO-CCSD(T)/cc-pVTZ is used to calculate the ground state, and the first 30 excited states of molecule in methanol were calculated by TD-DFT-B3LYP/cc-pVTZ level of theory in SMD solvent model. To analy the excited states of sulforaphane molecule by calculate UV spectrum and hole-electron. Finally, we predicted the active sites of sulforaphane molecule by calculating frontier orbital and fukui function. The results show that: In the UV spectrum, the absorption peaks at 204.4602 and 336.5590nm are the absorption bands of hetero atomic groups of conjugate molecules, which are generated by n-π* transition and belong to R absorption band. According to hole-electron analysis, S0→S18 is the local excitation, and S0→S7, S0→S25 and S0→S29 are the charge transfer excitation. In addition, the O and S atoms on sulfuryl group of sulforaphane are the site of electrophilic reaction; the C and S atoms on the cyanide group are sites of nucleophilic reaction. This study provides a theoretical basis for better understanding the antioxidant and anticancer mechanisms of sulforaphane.