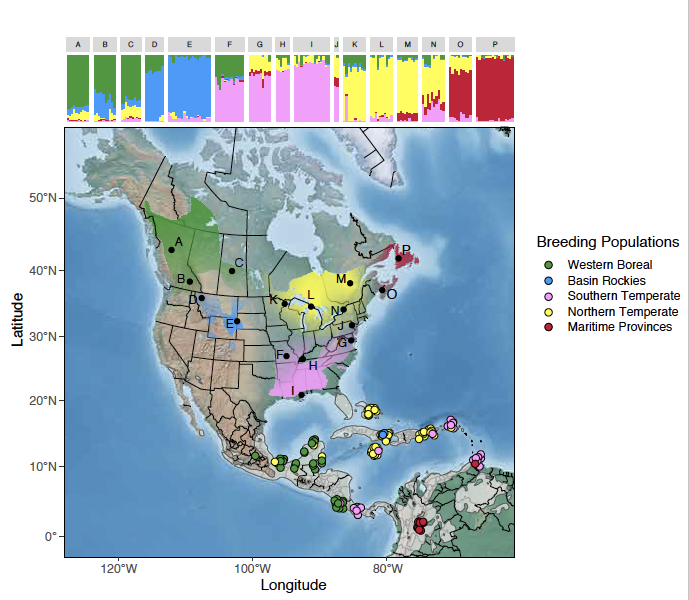
Understanding the geographic linkages among populations across the annual cycle is an essential component for understanding the ecology and evolution of migratory species and for facilitating their effective conservation. While genetic markers have been widely applied to describe migratory connections, the rapid development of new sequencing methods, such as low-coverage whole genome sequencing (lcWGS), provides new opportunities for improved estimates of migratory connectivity. Here, we use lcWGS to identify fine-scale population structure in a widespread songbird, the American Redstart (Setophaga ruticilla), and accurately assign individuals to genetically distinct breeding populations. Assignment of individuals from the nonbreeding range reveals population-specific patterns of varying migratory connectivity. By combining migratory connectivity results with demographic analysis of population abundance and trends, we consider full annual cycle conservation strategies for preserving numbers of individuals and genetic diversity. Notably, we highlight the importance of the Northern Temperate-Greater Antilles migratory population as containing the largest proportion of individuals in the species. Finally, we highlight valuable considerations for other population assignment studies aimed at using lcWGS. Our results have broad implications for improving our understanding of the ecology and evolution of migratory species through conservation genomics approaches.