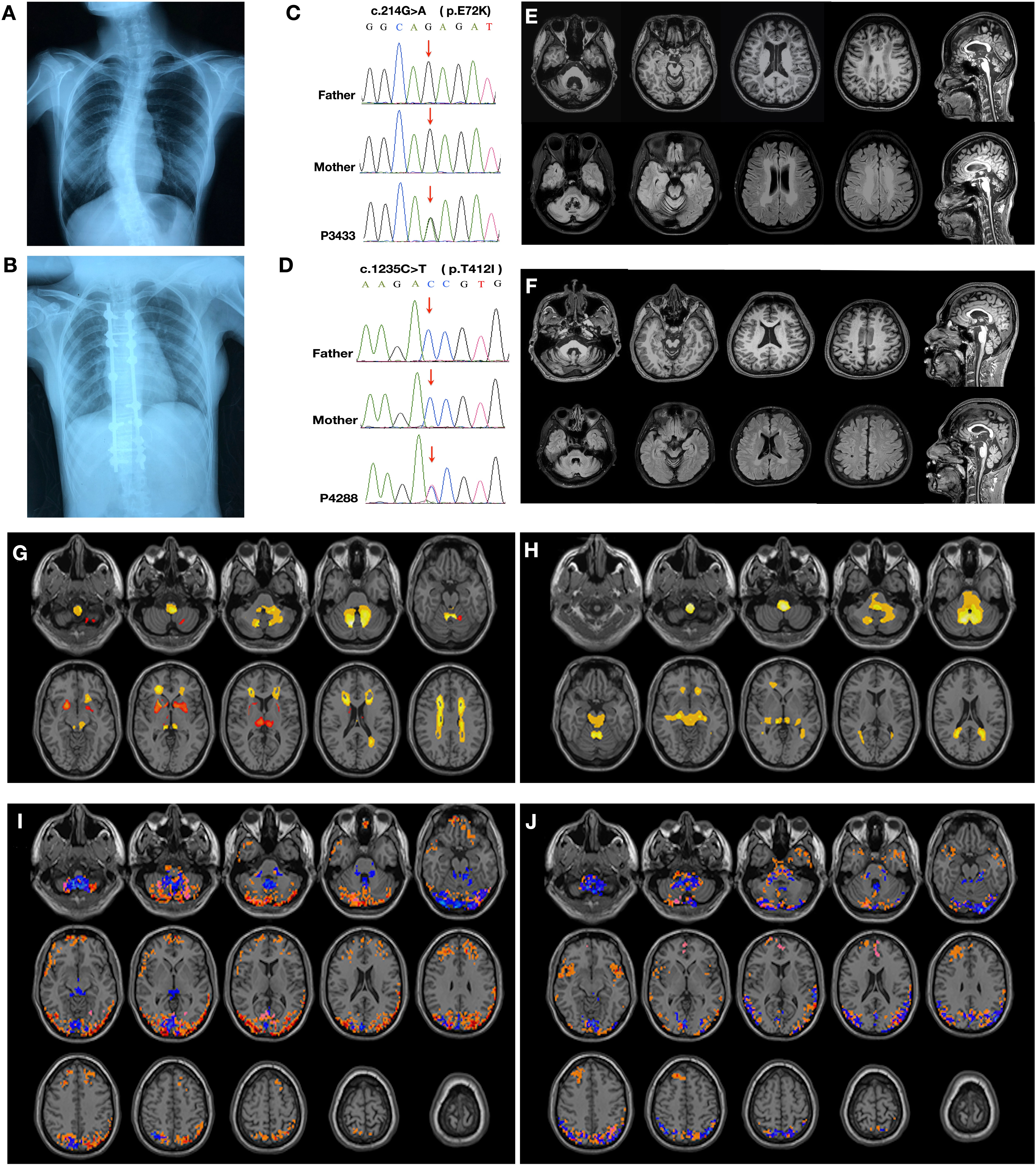
Two de novo mutations in GFAP gene were identified (c.214G>A, p.E72K and c.1235C>T, p.T412I) by whole exome sequencing. The common clinical features of the two patients was bulbar dysfunction, pyramidal signs and white matter lesions in periventricular regions. We conducted a novel data-driven method to explore the atrophic pattens and spontaneous brain functional network activity according to the neuroimaging data. Similar atrophic patterns, increased brain functional connectivity in occipital and posterior parietal cortex were detected in the two probands. Western blotting revealed the decreased level of GFAP with p.T412I mutation, while p.E72K and p.R239C mutations were at a similar level to wild type, suggesting the mutations located in the tail domain could decrease the solubility of GFAP. Abnormal inclusions of mutant GFAP were colocalized with ubiquitin, 20S proteasome, protein 1 light chain 3-II (LC3-II) and lysosome. The mutant GFAP caused activated autophagy flux while ubiquitin-proteasome pathway could be blocked as a mechanism for degrading aggregates.We herein presented two AxD patients with heterozygous mutations in GFAP. We noticed that mutant GFAP aggregations induced activated autophagy upon proteasome degrading pathway impairment. Our findings further expand the clinical and genetic spectrum of AxD.