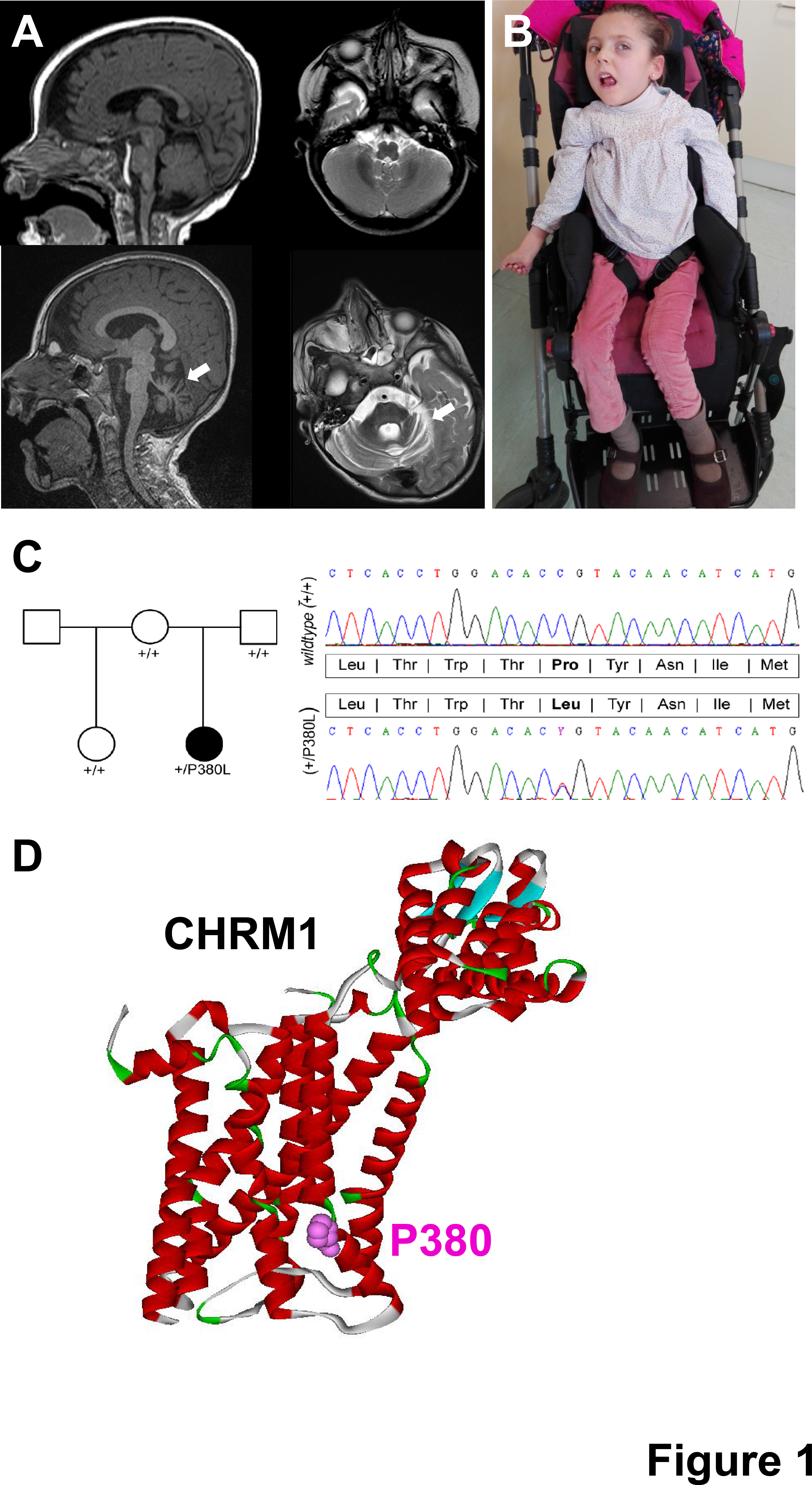
Developmental and epileptic encephalopathies are a group of devastating disorders where an underlying, usually genetic, cause leads to brain developmental impairment which can be further aggravated by superimposed, abundant epileptiform activity. Compared to other epilepsies, DEE show much greater locus heterogeneity and de novo rare damaging variants in genes involved in critical developmental pathways, notably regulation of synaptic transmission, have emerged as a frequent cause. Here, in a young girl with early-onset refractory epilepsy, severe disability and progressive cerebral and cerebellar atrophy, trio-based whole exome sequencing analysis uncovered a de novo missense variant in CHRM1. No additional CHRM1 variants were found by WES reanalysis in a cohort of 102 patients with EIEE/DEE nor any matches were produced upon sharing the novel variants in the Matchmaker Exchange platform. Biochemical analyses proved that this variant caused a reduction in protein levels and an impaired cellular trafficking. In addition, the mutated receptor showed defective activation of intracellular signalling pathways. Our data strengthen the concept that brain reduced muscarinic signalling lowers seizure threshold and severely impairs neurodevelopment.