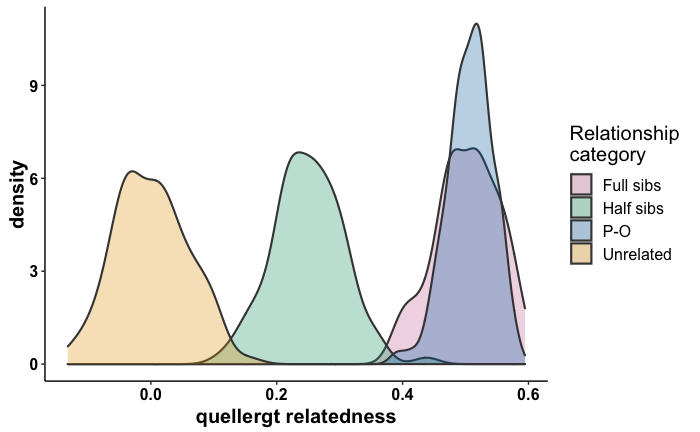
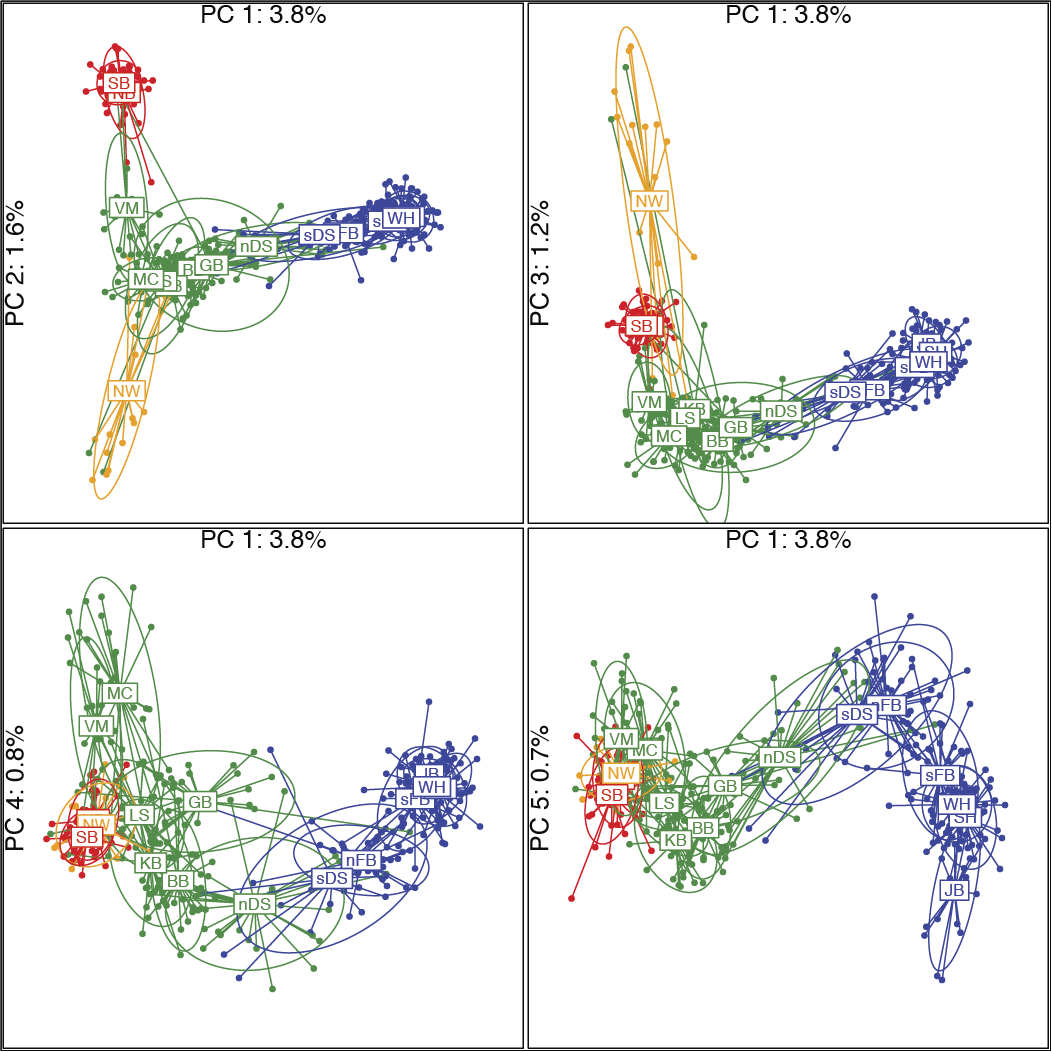
Single-nucleotide polymorphisms (SNPs) have numerous advantages over microsatellites, including greater power to infer population structure and history and to detect loci undergoing selection. Here, we conduct the first continental-level SNP study of polar bears (Ursus maritimus) using genotypes from an array of 5441 SNP loci genotyped in 16–30 polar bears sampled in each of 16 geographic regions in Canada and West Greenland. Our study aimed to assess population history and genetic structure and to identify evidence of adaptive loci. Using these data, we confirmed the existence of four broad-scale genetic clusters in North America (FCT = 0.035) and identified nine fine-scale subclusters using more powerful spatial methods. An assessment of historical patterns of migration suggests that polar bears migrated into North America from the Beaufort Sea after the last glacial maximum. Using a conservative approach, we identified 17 loci that may represent adaptive variation, including one SNP in the 3’ untranslated region of PDLIM5 (PDZ And LIM Domain 5), a gene involved in cardiovascular function, which has undergone substantial selection in polar bears since their divergence from brown bears. Outlier loci differentiated the Norwegian Bay genetic cluster more strongly from remaining clusters than did our complete dataset, suggesting possible adaptive differences in the High Arctic. Through careful consideration of SNP loci, sample inclusion, and analytical approaches, we provide a comprehensive picture of polar bear population structure at a continental level. This study provides a model for the analysis of wide-ranging species that can contribute to their conservation and management.