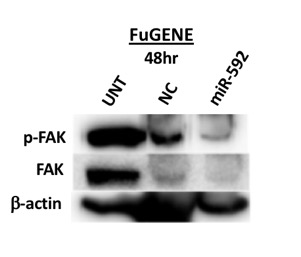
Abstract Triple-negative breast cancer (TNBC) is a disease characterized by its aggressive metastasis and poor prognosis. Developing targeted therapies that increase the tissue selectivity of drugs would be beneficial to the improvement of the overall efficacy of anti-cancer treatment and the suppression of the mortality rate of TNBC patients. The type 1 insulin-like growth factor receptor (IGF-1R) and focal adhesion kinase (FAK) pathways are involved in cell migration and cell invasion of the metastatic cascade in TNBC. The upregulation of miR-592 is has been shown to reduce cell proliferation and invasion in cancer cells, however, its role in TNBC remains unknown. It is demonstrated in this study that the overexpression of miR-592 resulted in decreased FAK expression, cell migration, and colony formation in the MDA-MB-231 cell line. Comprehensively, miR-592 is proposed as an important repressor of the development and progression of TNBC linked to epithelial-mesenchymal transition and tumorigenesis. Understanding the exact mechanism of action of miR-592 can reveal new therapeutic targets for the treatment of mesenchymal TNBCs. Introduction Triple-negative breast cancer (TNBC) involves the lack of the estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth receptor 2 (HER2) (1–3). This subtype exhibits aggressive metastasis and accounts for 10-20% of invasive breast cancers as of 2009 (3). The prevalence of TNBC and similar basal-like breast cancer subtypes is significantly higher in pre-menopausal African American and Latino women (4,5). The TNBC subtype is characterized by high risk of recurrence, distant metastasis and high mortality rates within 5 years (6). Current gene-targeted therapies, such as tamoxifen and Herceptin, target transmembrane receptors that the TNBC subtype does not possess (1). The use of chemotherapeutic drugs and radiation also has not displayed promising prevention of the poor prognosis of TNBC patients (7). Due to the lack of ER, PR, and HER2 receptors in TNBC, there still remains a great need for targeted gene therapy through the understanding of cellular mechanisms involves in the aggressive behavior of TNBC. The tyrosine kinase type 1 insulin-like growth factor receptor (IGF-1R) has been recognized for its role in tumor cell proliferation and increased cancer risk (8–10). Its ligand, the IGF-1 hormone, is abundant in human serum and released primarily from the liver. The upregulation of intracellular signaling of IGF-1R leads to the activation of the PI3K/AKT and MAPK pathways promoting cancer development and progression through maintenance of cell proliferation and survival (9,11,12). Significant crosstalks between IGF-1R and a variety of other pathways, including those involving mesenchymal-epithelial transition factor (MET) and vascular endothelial growth factor (VEGFR), been shown to promote the development and progression of various cancers (10). A particular crosstalk between the IGF-1R and focal adhesion kinase (FAK) signaling pathways have been shown to be involved in the regulation of migratory and invasive behaviors associated with metastasis in mesenchymal TNBCs (13). Due to its interaction in complex networks, IGF-1R has been noted as an interesting therapeutic target. microRNAs (miRNAs) are short single-stranded non-coding RNA involved in the post-transcriptional regulation (14). miRNAs negatively regulate gene expression through mechanisms involving translation repression by forming the RISC complex and binding to the 3’ untranslated region (UTR), preventing interaction of polyadenylate-binding protein (PABP) on the 3’ UTR with the 5’ UTR that is required for ribosomal assembly or mRNA degradation by perfect complementary binding to mRNA (15,16). Studies have recently displayed that tumor suppressor or oncogenic miRNAs interact in pathways involving metastasis, apoptosis, and cell proliferation (14,17,18). As a result of its attractive potential therapeutic function, recent studies have analyzed the effect of expression of miRNA in a variety of cancers as they are located in cancer-linked genomic regions and involved in cancer-related pathways (18–20). IGF-1R has been shown to be a direct target gene for microRNA-592 in hepatocellular carcinoma cells and the upregulation of the miR-592 resulted in decreased cell proliferation and cell invasion (20). The upregulation of microRNA-592 yielded significantly reduced levels of cell proliferation and invasion in lung cancer, hepatocellular carcinoma, and in gliomas (10,20–22). However, miRNA-592 regulation has not been extensively studied in breast cancer cells, especially in triple negative breast cancer cells. A recent study investigated the overexpression of miRNA-592 in breast cancer cell lines which resulted in downregulation of TGFβ-2, a direct target oncogene of miRNA-592 (23). miRNA profiling of TNBC cells in Latin American patients also revealed that miRNA-592 is expressed in TNBC cell lines and could be involved in cancer-related pathways (24). It was hypothesized that the overexpression of miRNA 592 will inhibit the migration and cell growth of TNBC cancer cells by affecting the signaling of IGF-1R and FAK. This current study displays the connection with the expression of miRNA-592 and the regulation of the FAK pathway in TNBC cells through immunoblotting, wound healing assay, and clonogenicity assay. The results of this study demonstrate that miRNA-592 could be a worthy candidate for investigation in TNBC cell lines as a possible tumor suppressor. Materials and Methods Reagents Radioimmunoprecipitation assay (RIPA) cell lysis buffer was from Cell Signaling Technology, Inc. (Beverly, MA, USA), Nonidet P-40 (NP40) cell lysis buffer was from Boston BioProducts (Ashland, MA, USA), phenylmethylsulfonyl fluoride (PMSF) was from NOVUS (Littleton, CO, USA), and EZ Block protease and phosphatase inhibitor cocktail was purchased from Bio Vision (San Francisco, CA, USA). Phosphate buffer saline (PBS) was from Mediatech (Manassas, VA, USA). Cell Culture MDA-MB-231 cell line was obtained from American Type Culture Collection (Manassas, VA, USA). This cell line was authenticated. Cells were routinely maintained in complete media, including Dulbecco’s Modification of Eagle’s Medium (DMEM) (CORNING) supplemented with 10% Fetal Bovine Serum (FBS) (CORNING) and 2M L-glutamine (Invitrogen, Carlsbad, CA, USA). Cells were cultured in humidified incubators at 37°C with 5% CO2. miR-592 Transfection miR-592 Mimic (HMI0001-HMI2785) was a small, double-stranded RNA molecule utilized to imitate endogenous mature miR-592 molecules. miR-592 Mimic was commercially available from Sigma-Aldrich (St. Louis, MO, USA). A scrambled sequence miRNA (miR-NC) (HMC0002) was also used as a negative control (Sigma-Aldrich). To rehydrate the miR-592 and miR-NC, each pellet was mixed with 250L of Molecular Grade Water from G-Biosciences (St. Louis, MO, USA) to produce 20M stocks. INTERFERin transfection reagent was obtained from Polyplus (New York, NY, USA). FuGENE transfection reagent was obtained from Promega (Madison, WI, USA). Both reagents were utilized to optimize miRNA transfection according to the manufacturer’s instruction. miR-592 and miR-NC were gently mixed with INTERFERin or FuGENE in serum-free DMEM and incubated at room temperature for 10 minutes for complex formation. MDA-MB-231 cells were plated and grown to 70-80% confluency. After incubation, the complex was added to the plated cells. The untreated cells contained complete media only. Forty-eight hours post-transfection, cells were subsequently used in Western blotting, wound-healing assay, and clonogenicity assay as detailed below. Western Blotting Following 48-72 hours treatments, MDA-MB-231 cells were washed with PBS and harvested using RIPA or NP40 lysis buffer containing PMSF and protease/phosphatase inhibitors according to the manufacturer's protocol. The total protein concentration of each treatment following transfection was measured utilizing the Bradford Assay (Bio Rad; Hercules, CA, USA). Equal amounts of protein, 20 µg, for each sample was separated via SDS-PAGE at 80V for 20 minutes and 125 V for 45 minutes. The polyvinylidene difluoride (PVDF) membranes (Bio Rad) were activated with methanol for 5 minutes. The separated proteins were transferred to either methanol-activated PVDF or nitrocellulose membranes (Bio Rad) overnight at 4C at 23V. The antibody against -actin, an internal loading control, was purchased from NOVUS (Littleton, CO, USA) were used for Western Blots according to standard protocol. Primary antibodies against IGF1R, anti-pIGF-1R were from Cell Signaling Technologies, Inc. (Danvers, MA, USA). Primary antibodies anti-FAK, anti-p-FAK were from BD Biosciences (San Jose, CA, USA). The goat anti-rabbit and goat anti-mouse secondary antibodies were from SouthernBiotech (Birmingham, AL, USA). Following a blocking incubation with 5% non-fat milk at room temperature for 2 hours, the membranes were incubated with rocking at 4˚C overnight with primary anti IGF 1R, anti-pIGF-1R (dilution, 1:1,000) and anti-FAK, anti-p-FAK (dilution, 1:1,000), followed by incubation at room temperature for 1 hour with the goat anti-rabbit or goat anti-mouse horseradish peroxidase-conjugated secondary antibody (1:5,000-10,000 dilution). The proteins were developed with Luminata Classico Western horseradish peroxidase substrate from Millipore (Billerica, MA, USA) and visualized using the c300 Azure imaging system (Dublin, CA, USA). Wound-Healing Assay Cells were seeded in a 24-well and transfected at 80-90% confluency. At 100% confluency, the monolayer cells were wounded by scratching with a sterilized 200uL pipette tip, ensuring the width of each wound was the same. The wells were rinsed with PBS three times to remove floating cells and debris. Complete media was added, and wounds were immediately imaged (0hr time point). Cells were then incubated for 24 hours at 37°C. Pictures were acquired using a Motic AE31 microscope (Carlsbad, CA, USA), moticam 2500 digital camera from Motic software (Carlsbad, CA, USA) at indicated time (0hr, 24hr, and 48hr). Clonogenicity Assay Single cell suspensions were seeded in a 6-well plate at a density of 250 cells per well and allowed to grow. The following day, cells will be transfected with miR-592 or miR-NC as described above. After a 6-day treatment period, the media was removed. Cells were fixed in 10% ice-cold methanol, stained with crystal violet (0.1% in 20% methanol), and allowed to dry overnight. Colony numbers will be assessed visually and colonies containing >25 normal appearing cells were counted. Pictures were acquired using a Motic AE31 microscope (Carlsbad, CA, USA), moticam 2500 digital camera from Motic software (Carlsbad, CA, USA). Statistical Analysis For the clonogenicity assay results, the average colonies observed for each treatment and standard error bars produced were determined utilizing Microsoft Excel. Results miR-592 overexpression inhibits FAK but not IGF-1R expression in TNBC cells The crosstalk between the IGF-1R and FAK pathways have been shown to be associated with the metastasis of TNBC tumor cell (13). To obtain evidence of the effects of overexpression of miR-592 on FAK expression in cultured TNBC cells, FAK protein levels were determined by Western blotting in the MDA-MB-231 cell line (Figure 1). Without treatment, MDA-MB-231 cells expressed high levels of p-FAK and FAK protein. The introduction of miR-NC into TNBC with FuGENE for 48 hours resulted in less p-FAK protein expression than in the untreated cells. This negative control also caused much lower expression of FAK protein (Figure 1). As expected, following the transfection of miR-592 with the FuGENE reagent for 48 hours, the TNBC cells low levels of p-FAK and FAK, specifically undetectable levels for FAK protein.