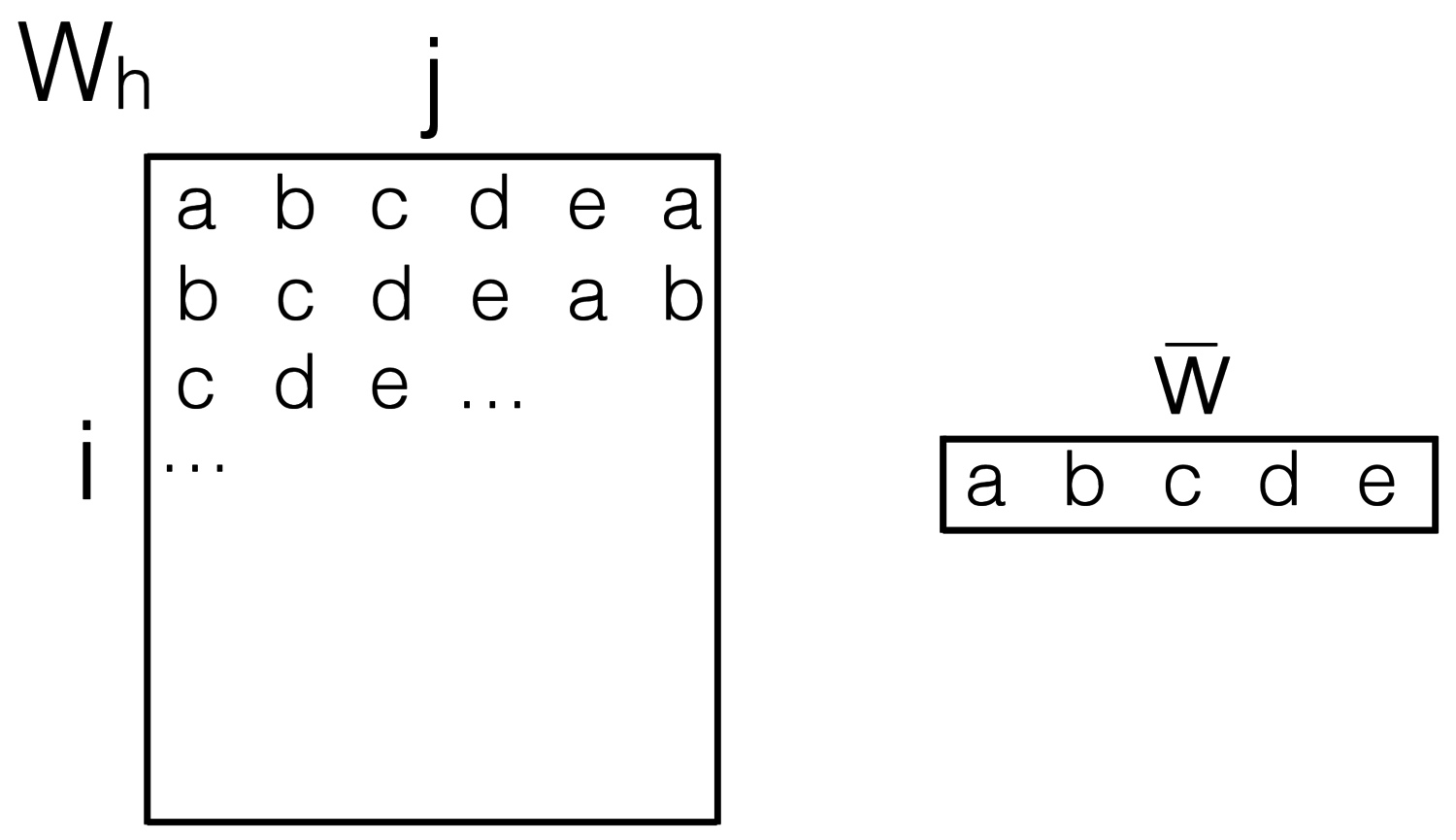
INTRODUCTION As available datasets increase in size, machine learning models can successfully use more and more parameters. In applications such as computer vision, models with up to 144 million parameters are not uncommon and reach state-of-the-art performance. Experts can train and deploy such models on large machines, but effective use of lower-resource hardware such as commodity laptops or even mobile phones remains a challenge. One way to address the challenge of large models is through model compression using hashing . In general, this amounts to reducing a parameter set S = {s₀, s₁, ...sD} to a greatly reduced set R = {r₀, r₁, ..., rd} with d ≪ D by randomly tying parameters to hash buckets (si = rh(i)). This turned out to perform very well for neural networks, leading to the so-called HashedNets. Many machine learning models involve several linear projections representable by matrix-vector products W ⋅ x where x is input data and W consists of model parameters. In most such models, this linear algebra operation is the performance bottleneck; neural networks, in particular, chain a large number of matrix-vector products, intertwined with non-linearities. In terms of dimensionality, modern systems deal with millions training samples xi lying in possibly high-dimensional spaces. The shape of W, (dout, din), depends on how deep a layer is in a network: at the first layer, din depends on the data being processed, while dout at the final layer depends on the desired system output (i.e., dout = 1 for binary classification, and dout = p if the output can fall in p classes). In middle layers, dimensionality is up to the model designer, and increasing it can make the model more powerful but bigger and slower. Notably, middle layers often have square Wh. When W is stored in a reduced hashed format Wh, many common trade-offs may change. The goal of our project is to explore the performance bottlenecks of the Wh ⋅ x operation where Wh is a hashed representation of an array that stays constant for many inputs xi. Since neural networks are typically trained with batches of input vectors x concatenated into an input matrix X, we will look at the general case of matrix-matrix multiplication, where the left matrix is in a reduced hashed format Wh ⋅ X. Taking advantage of massively parallel GPU architecture can be important even when dealing with smaller models. In March 2015, Nvidia announced a SoC for mobile devices with a GPU performance of 1 teraflop, the Tegra X1 ; we foresee future mobile devices to have stronger and stronger GPUs. The objectives of our project are to: 1. Investigate fast applications of Wh ⋅ X when Wh is small enough to be fully loaded into memory. In this case, is it faster to first materialize the hashed array and use existing fast linear algebra routines? Can the product be computed faster on a GPU with minimal memory overhead? This can lead to highly efficient deployment of powerful models on commodity hardware or phones. 2. Analyze performance when even after hashing Wh is too big. In the seminal work that popularized the usage large scale deep convolutional neural networks and training using the GPU , Krizhevsky predicts that GPUs with more memory can lead to bigger networks with better performance. Hashing-based compression can help practitioners prototype very large models on their laptops before deciding which configuration to spend cloud computing resources on. Can we make HashedNets training on GPUs efficient? This may involve forcing a predictable split on the hash function to allow for independent division of work.