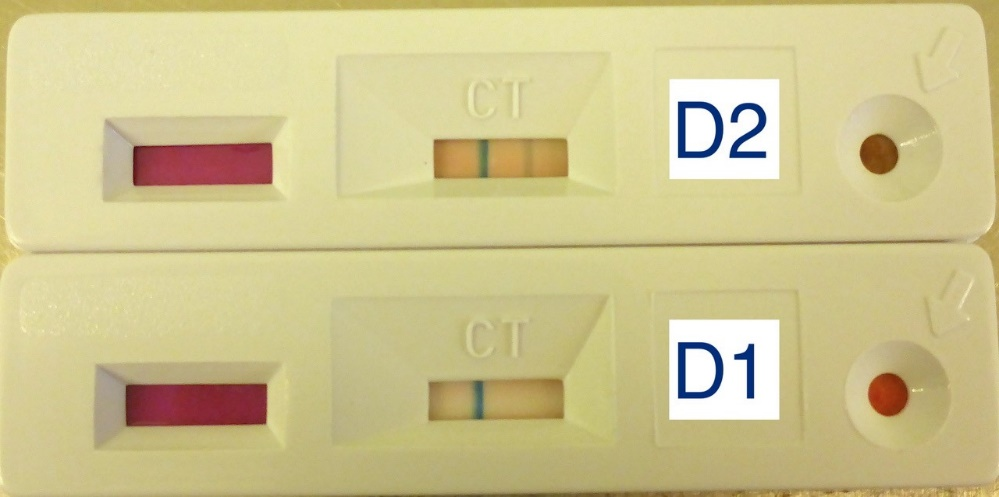
Porcine circoviruses are important pathogens of production swine. Porcine circovirus type 1 (PCV1) is non-pathogenic, and discovered as a contaminant of a porcine kidney cell line, PK-15. The discovery of pathogenic variant, PCV2, occurred in the late 90’s in association with post-weaning multi-systemic wasting disease syndrome (PMWS), which is characterized by wasting, respiratory signs and lymphadenopathy in weanling pigs. A new PCV type, designated as PCV3, was discovered in 2016, in pigs manifesting porcine dermatitis and nephropathy syndrome (PDNS), respiratory distress and reproductive failure. Pathological manifestations of PCV3 Infections include systemic inflammation, vasculitis and myocarditis. A 4 th PCV type, PCV4, was identified in 2020 in pigs with PDNS, respiratory and enteric signs. All the pathogenic PCV types are detected in both healthy and morbid pigs. They cause chronic, systemic infections with various clinical manifestations. Dysregulation of the immune system homeostasis is a pivotal trigger for pathogenesis in porcine circoviral infections. While the study of PCV3 immunobiology is still in its infancy lessons learned from PCV2 and other circular replication-associated protein (Rep)-encoding single stranded(ss) (CRESS) DNA viruses can inform the field of exploration for PCV3. Viral interactions with the innate immune system, interference with dendritic cell function coupled with the direct loss of lymphocytes compromises both innate and adaptive immunity in PCV2 infections. Dysregulated immune responses leading to the establishment of a pro-inflammatory state, immune complex associated hypersensitivity, and the necrosis of lymphocytes and immune cells are key features of PCV3 immunopathogenesis. A critical overview of the comparative immunopathology of PCV2 and PCV3/4, and directions for future research in the field are presented in this review.