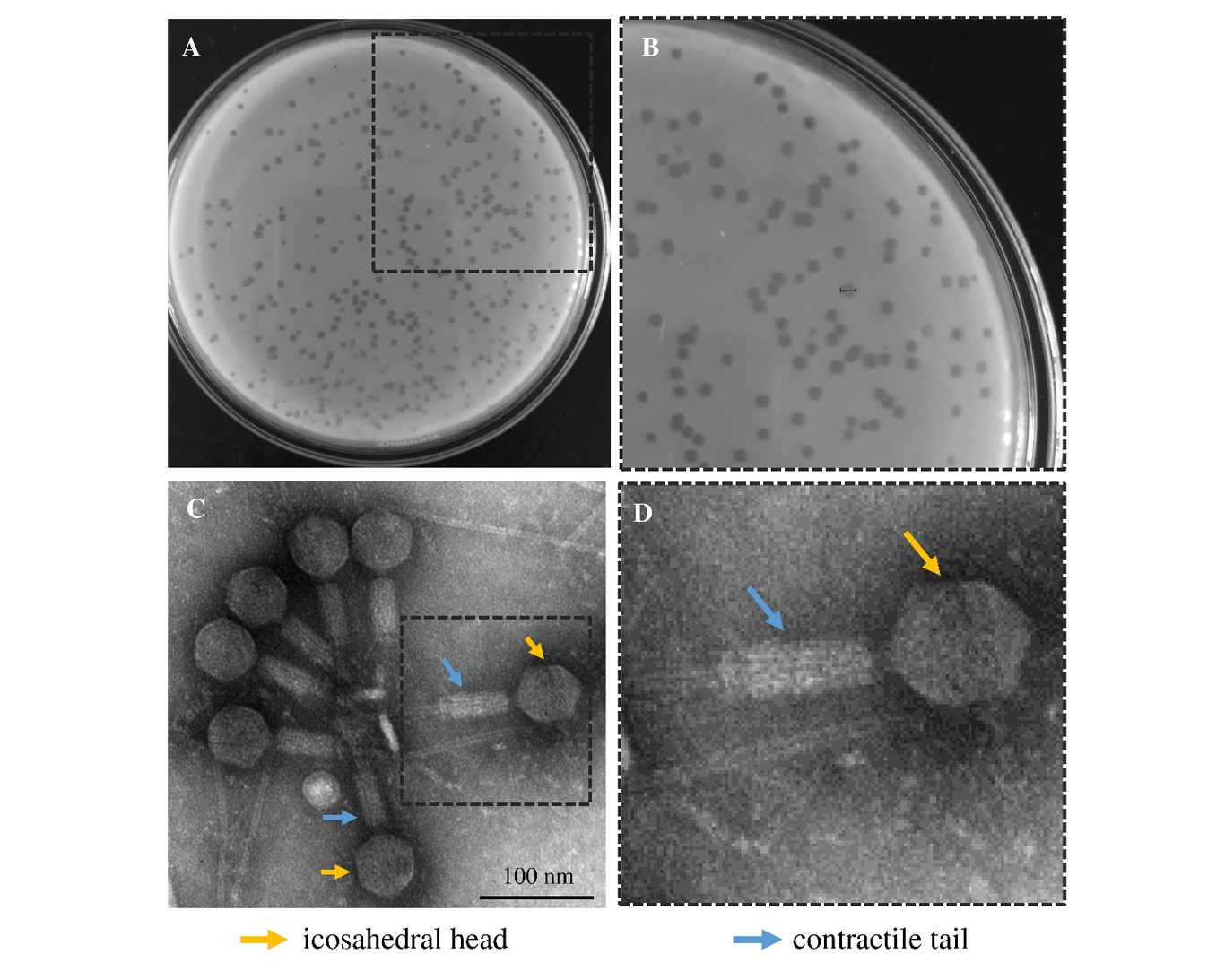
Introduction: The Coronavirus Disease 2019 (COVID-19) pandemic has had a variable worldwide impact, likely related to country-level characteristics. In this ecological study, we explored the association of COVID-19 case rates (per 100,000 people) and death rates (per 100,000 people) with country-level population health characteristics, economic and human development indicators, and habitat-related variables. Methods: To calculate country-level COVID-19 case and death rates, the number of cases and deaths were extracted from the Johns Hopkins Coronavirus Resource Center for 2020. Country-level population health characteristics, economic and human development indicators, and habitat-related variables were extracted from several publicly available online sources of international organizations. Results were tabulated according to world zones and country economies. Univariate and multivariable linear regression analyses were performed to examine determinants of COVID-19 case rates and death rates. Results: A total of 187 countries and territories were analyzed, with an aggregate COVID-19 case rate of 779 per 100,000 people, a death rate of 19 per 100,000 people, and a case-fatality rate of 2.4%. For country-level population health characteristics, a higher percentage rate of adults with obesity and a higher percentage rate of adults with high blood pressure was independently associated with a higher COVID-19 case rate, and a higher percentage rate of adults with obesity was associated with a higher COVID-19 death rate. For country-level economic and human development indicators, only a higher gross domestic product percentage rate spent on total health expenditure and a higher human development index was independently associated with a higher COVID-19 case rate and death rate. A higher percentage of urban population was independently associated with a higher COVID-19 death rate, whereas a higher income per capita was independently associated with a lower COVID-19 death rate. For country-level habitat-related variables, a higher average household size and a higher percentage rate of population with primary reliance on polluting fuels and technologies was independently associated with a lower COVID-19 case rate and death rate whereas a higher percentage rate of households with at least one-member age 65 years or over was associated with a higher case rate and death rates. Conclusion: This ecological study informs the need to develop country-specific public health interventions to better target populations at high risk for COVID-19, and test environmental interventions to prevent indoor transmission of SARS-CoV-2, taking into consideration population health characteristics, economic and human development indicators, and habitat-related variables that are unique to each country.