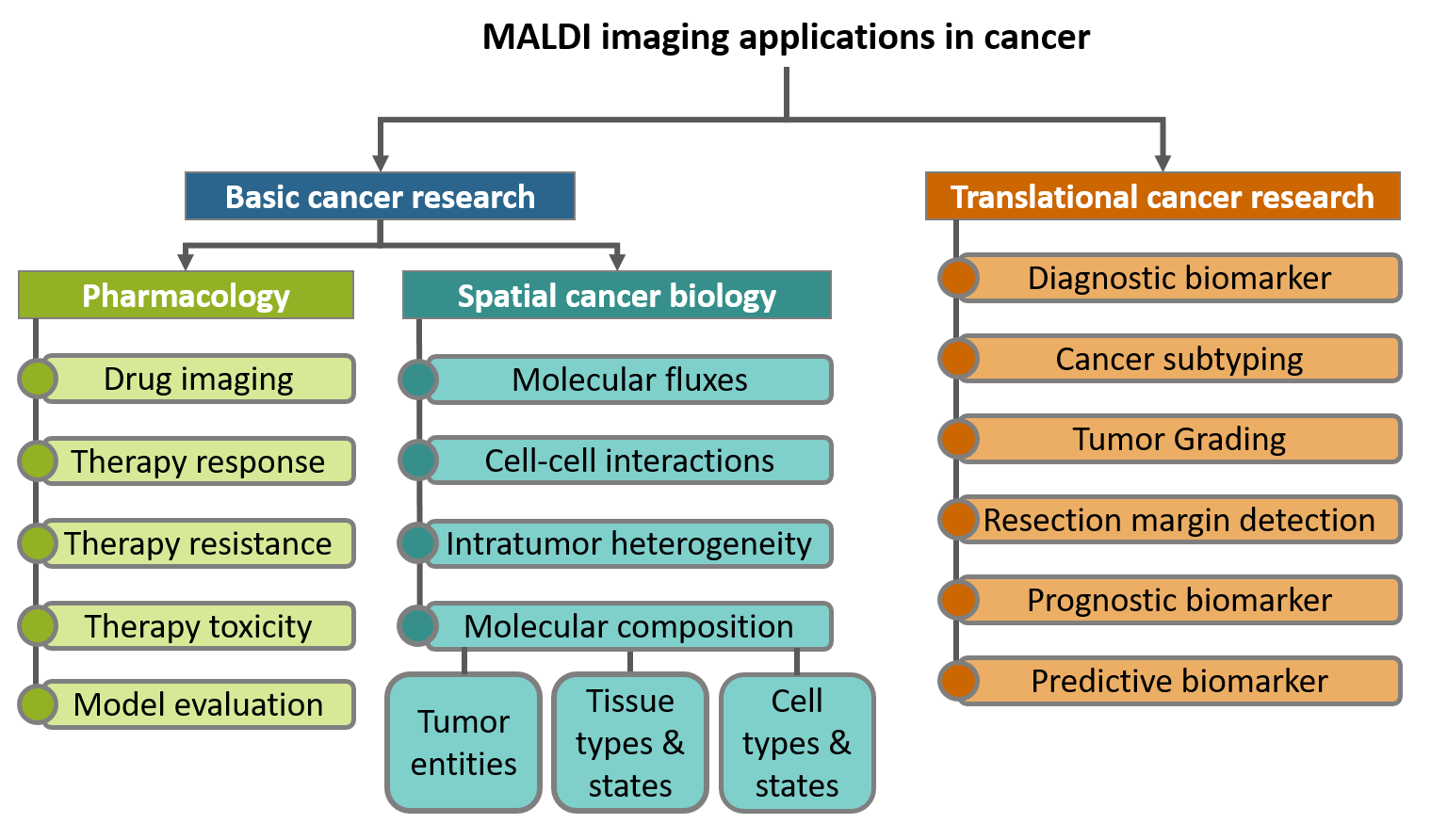
MALDI mass spectrometry imaging (MALDI imaging) is uniquely suited to advance cancer research by measuring spatial distribution of endogenous and exogenous molecules directly from thin tissue sections. These molecular maps provide valuable insights into various aspects of basic and translational cancer research, including spatial tumor and tumor microenvironment biology, pharmacological interventions, and patient stratification. However, despite these advantages, the utilization of MALDI imaging in studying rare cancers, which comprise approximately 20% of all cancers, remains limited. Rare cancers pose unique challenges in medical research, resulting in understudied entities with suboptimal management and outcomes. In this review, we explore the value of MALDI imaging in sarcoma, as an example of a highly heterogeneous and challenging rare cancer. We summarize existing MALDI imaging studies in sarcoma and outline potential future applications. In addition, we address the specific challenges encountered when employing MALDI imaging to rare cancers, and propose solutions, including the utilization of formalin-fixed paraffin-embedded tissues, multi-site studies, implementation of multiplexed experiments, and considerations for data sharing practices. Through this review, we aim to inspire collaboration between MALDI imaging researchers and clinical colleagues, to deploy the unique capabilities of MALDI imaging in rare cancer research, particularly in the context of sarcoma.