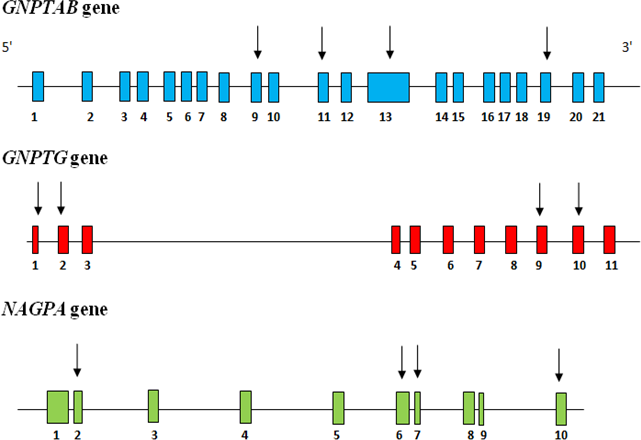
Stuttering is a childhood onset fluency disorder, intertwined with physiological, emotional and anxiety factors. The present study was designed to evaluate the recurrence of the reported mutations among three previously implicated (GNPTAB, GNPTG, NAGPA) candidate genes, in persons with stuttering (PWS) from south India. Mutation screening was performed on 64 probands on 12 specific exons, by Sanger sequencing. A total of 12 variants were identified, which included five nonsynonymous missense, five synonymous and two non coding variants. Only three unrelated probands, harbored heterozygous likely pathogenic missense variants (c.3598G>A in GNPTAB, c.802A>C in GNPTG and c.131G>C in NAGPA) resulting in an overall frequency of 4.7% and an allele frequency of 2.3% (3/128*100). Among the three likely pathogenic variants only two co-segregated (c.3598G>A in GNPTAB - STU 29 and c.802A>C in GNPTG - STU 63) with the affected status reducing the likely pathogenic allele frequency to 1.6% (2/128*100). The recurrence of pathogenic variants in our study corroborate the causative role of these genes in stuttering but still remains unknown as to how the speech dysfluency occurs even in its heterozygous condition. Keywords: Stuttering, candidate genes, GNPTAB, GNPTG, NAGPA