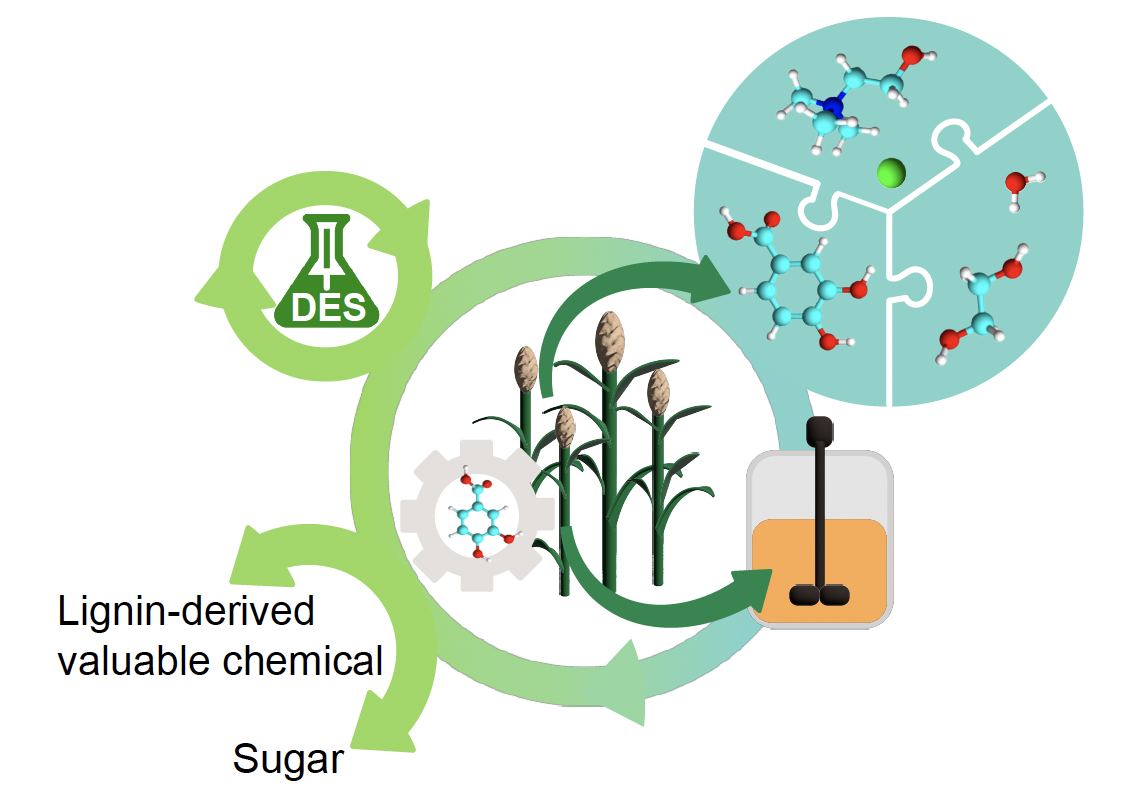
Biomass-derived deep eutectic solvents (DESs) have been introduced as promising pretreatment and fractionation solvents because of their mild processing conditions, easy synthesis, and green solvent components from biomass. In recent DES studies, solvent-based third constituents like water, ethanol, and others improve the processibility of typical binary DESs. However, the impacts of these components are not well understood. Here, two solvent-based constitutions, including water and ethylene glycol, were applied to 3,4-dihydroxybenzoic acid (DHBA)-based DES system for improving the conversion efficiency of cellulose-rich fraction and the properties of lignin fraction. Compositional changes, enzymatic digestibility of carbohydrate components, and transformation of lignin were used to evaluate the impact of each constituent on biomass processing. Ternary DHBA-ChCl DESs exhibited better performances in delignification, fermentable sugar production, and preservation of β–O–4 ether linkage in lignin compared to neat ChCl-DHBA DES.