AUTHOREA
Log in
Sign Up
Browse Preprints
LOG IN
SIGN UP
Jenna M. Lang
Postdoctoral fellow
Genome Center
Member of:
University of California, Davis
Public Documents
2
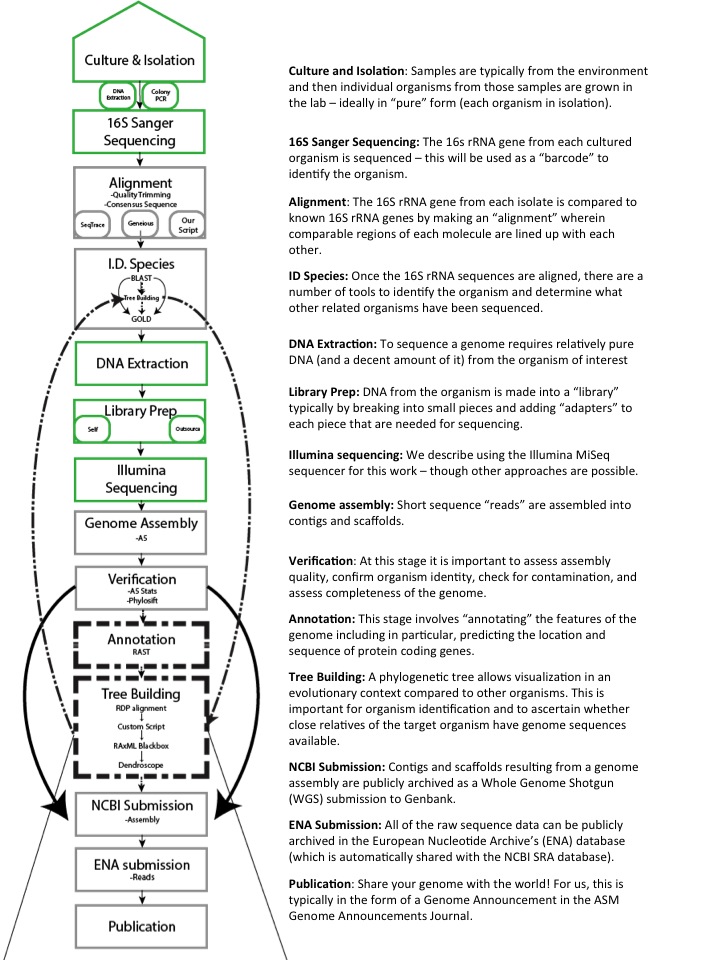
Swabs to Genomes: A Comprehensive Workflow
Jenna M. Lang, Guillaume Jospin, Aaron Darling, et al.
The Microbes We Eat
Jenna M. Lang, Jonathan A. Eisen, Angela M. Zivkovic, et al.