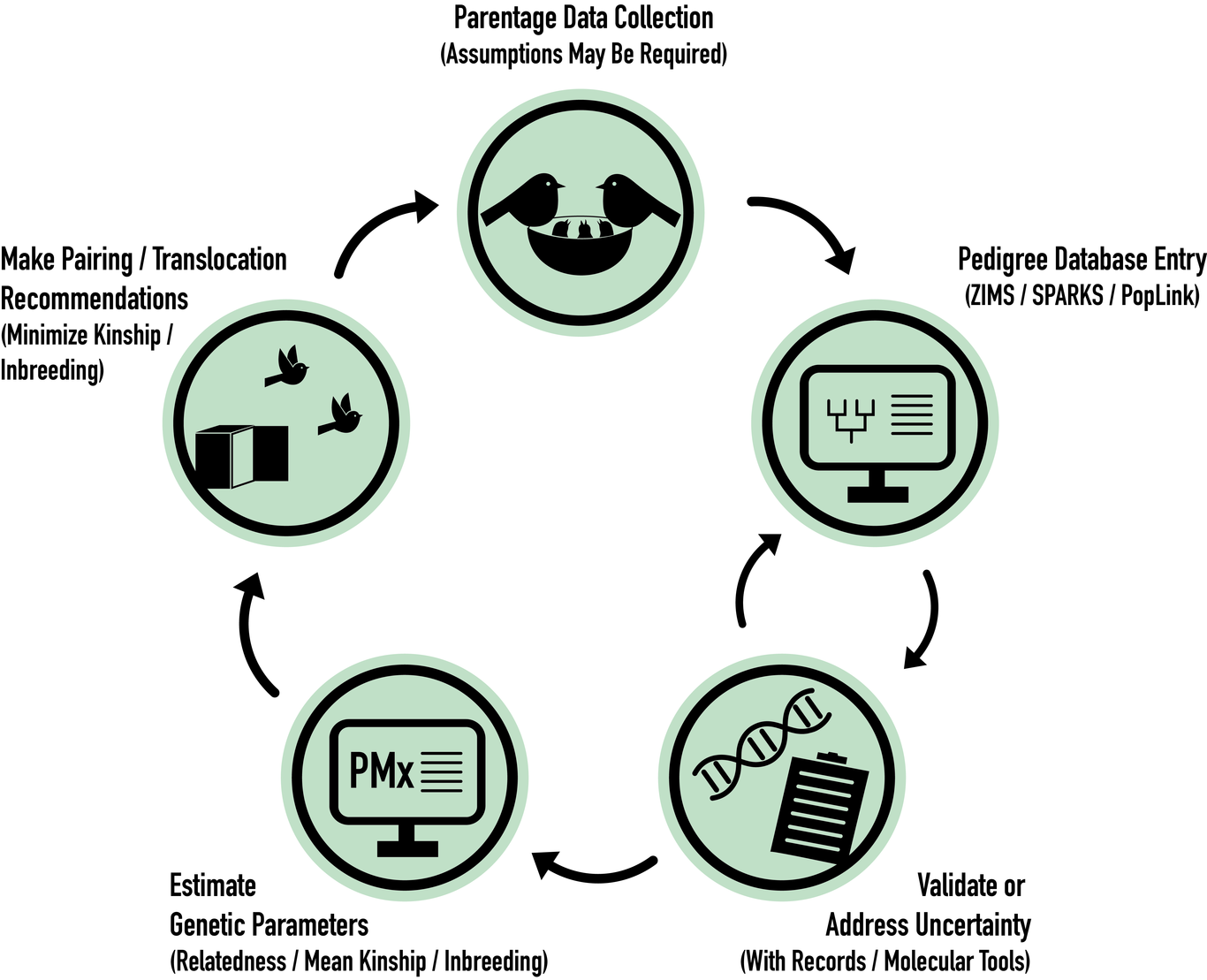
Over the past 50 years conservation genetics has developed a substantive toolbox to inform species management. One of the most long-standing tools available to manage genetics - the pedigree - has been widely used to characterize diversity and maximize evolutionary potential in threatened populations. Now, with the ability to use high throughput sequencing (HTS) to estimate relatedness, inbreeding, and genome-wide functional diversity, some have asked whether it is warranted for conservation biologists to continue collecting and collating pedigrees for species management. In this perspective, we argue that pedigrees remain a relevant tool, and when combined with genomic data, create an invaluable resource for conservation genomic management. Genomic data can address pedigree pitfalls (e.g., founder relatedness, missing data, uncertainty), and in return robust pedigrees allow for more nuanced research design, including well-informed sampling strategies and quantitative analyses (e.g., heritability, linkage) to better inform genomic inquiry. We further contend that building and maintaining pedigrees provides an opportunity to strengthen trusted relationships among conservation researchers, practitioners, Indigenous Peoples, and local communities. Keywords: conservation genomics, quantitative genetics, pedigree, kinship,ex situ , in situ