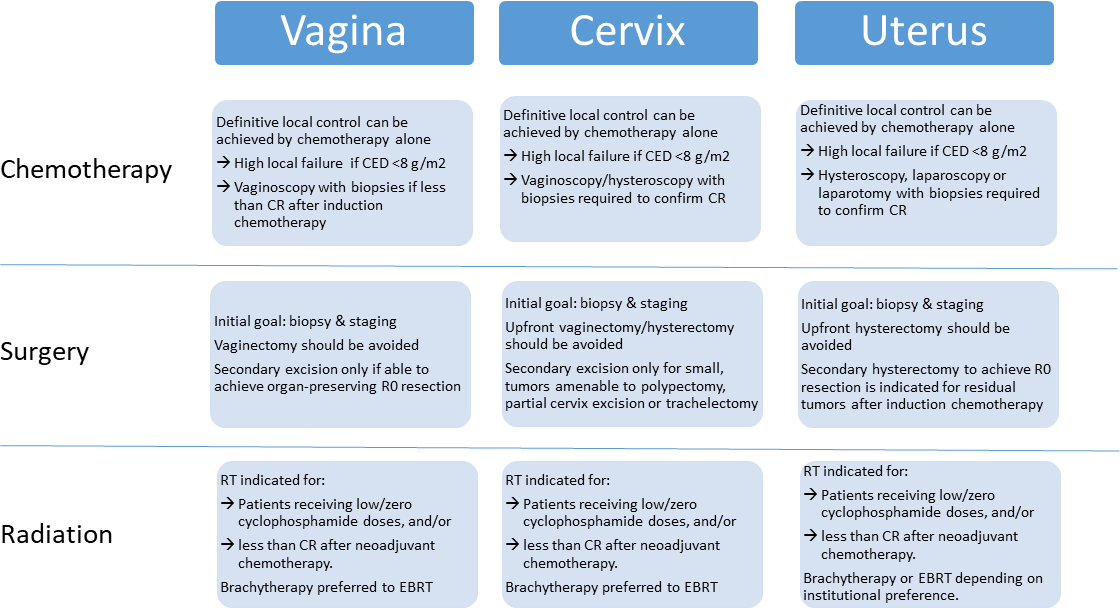
Pediatric oncology is justifiably proud of its long tradition of multi-institutional collaboration in clinical research. Perhaps no other field of medicine has more effectively shown what can be achieved by pooling talent and resources to study challenging diseases. Historically, most collaborative projects were limited to a single country or continent. However, more progress comes from even broader international cooperation. With rare cancers, this may be the only way to gather sufficient patient numbers to address key questions. Sharing national experiences can also lead to a deeper understanding of the advantages and risks associated with different therapeutic approaches. The first steps to increased global cooperation, however, is agreeing on a common language to describe patient cohorts and consensus standards to guide diagnosis, evaluation, and treatment. Applying these lofting goals to pediatric soft tissue sarcomas, the INternational Soft Tissue SaRcoma ConsorTium (INSTRuCT) was born.From its initial formative meeting in May 2017, INSTRuCT has patterned its structure and purpose on the successful model of the International Neuroblastoma Risk Group (INRG).1,2 The membership of INSTRuCT is composed of three large cooperative clinical trials organizations: Children’s Oncology Group (COG), Cooperative Weichteilsarkom Studiengruppe (CWS), and European paediatric Soft tissue sarcoma Study Group (EpSSG). The first goal for INSTRuCT is to develop an international risk stratification system for rhabdomyosarcoma (RMS) to replace competing systems used in Europe and North America. A common RMS risk stratification system would facilitate the comparison of clinical trial results across cooperative groups. Before generating a RMS risk stratification system, INSTRuCT agreed upon a standard RMS data dictionary, leveraging the University of Chicago’s Pediatric Cancer Data Commons expertise in data standardization.3 The compilation of COG, CWS, and EpSSG data (and data from their legacy groups) from finished studies into a single INSTRuCT dataset is nearly complete, and will include more than 7000 patients enrolled on previous RMS clinical trials. Once the RMS risk stratification project is finished, INSTRuCT will mine its dataset for answers to questions that can only be addressed with large, well-annotated clinical data. Future work will also include expanding the RMS data dictionary and adding a non-RMS soft tissue sarcoma dataset, also drawn from COG, CWS, and EpSSG clinical trials.As the multi-disciplinary members of INSTRuCT were defining their RMS data dictionary, they realized they had the opportunity to develop international consensus statements on the diagnosis, evaluation, and management of pediatric soft tissue sarcomas. Clinical trial protocols include guidelines for pathologic diagnosis, imaging staging evaluation, and local control approaches with surgery and radiation therapy varied by primary anatomic site. INSTRuCT provided the forum for international discussion and consensus building, with the goal of publishing these expert opinions for broad dissemination and use by pediatric oncologists, surgeons, radiation oncologists, radiologists, and pathologists worldwide. In this issue of Pediatric Blood & Cancer, Morris et al. publish the one of first in a series of consensus statements from INSTRuCT, focusing on the surgical management in the diagnosis and local control of RMS arising in the extremity.4 Morris et al. outline recommended biopsy approaches, the rational for routine use of regional lymph node evaluation including the role of fluorodeoxyglucose positron emission tomography, and the decision-making behind up-front versus delayed primary excision. The recommendations are guided by the principles of maximizing oncologic outcome while maintaining extremity function. Given the rarity of extremity RMS and the absence of randomized trials comparing different management strategies, Morris et al. draw upon a combination of clinical data and expert opinion in their consensus guidelines, carefully documenting the level of evidence that supports each of their recommendations. Nonetheless, these guidelines represent the current state of the art for surgical management of extremity RMS and the basis for future clinical trial recommendations. A similar consensus statement on RMS of the female genital tract has also been published in Pediatric Blood & Cancer, by Lautz et al.5 With these two consensus statements, the INSTRuCTPediatric Blood & Cancer special series is off to an excellent start, with manuscripts on the surgical management for other primary sites and the pathologic evaluation of RMS to follow soon. As INSTRuCT co-chairs, we are pleased to introduce INSTRuCT to the global pediatric oncology community and look forward to many more contributions to come.