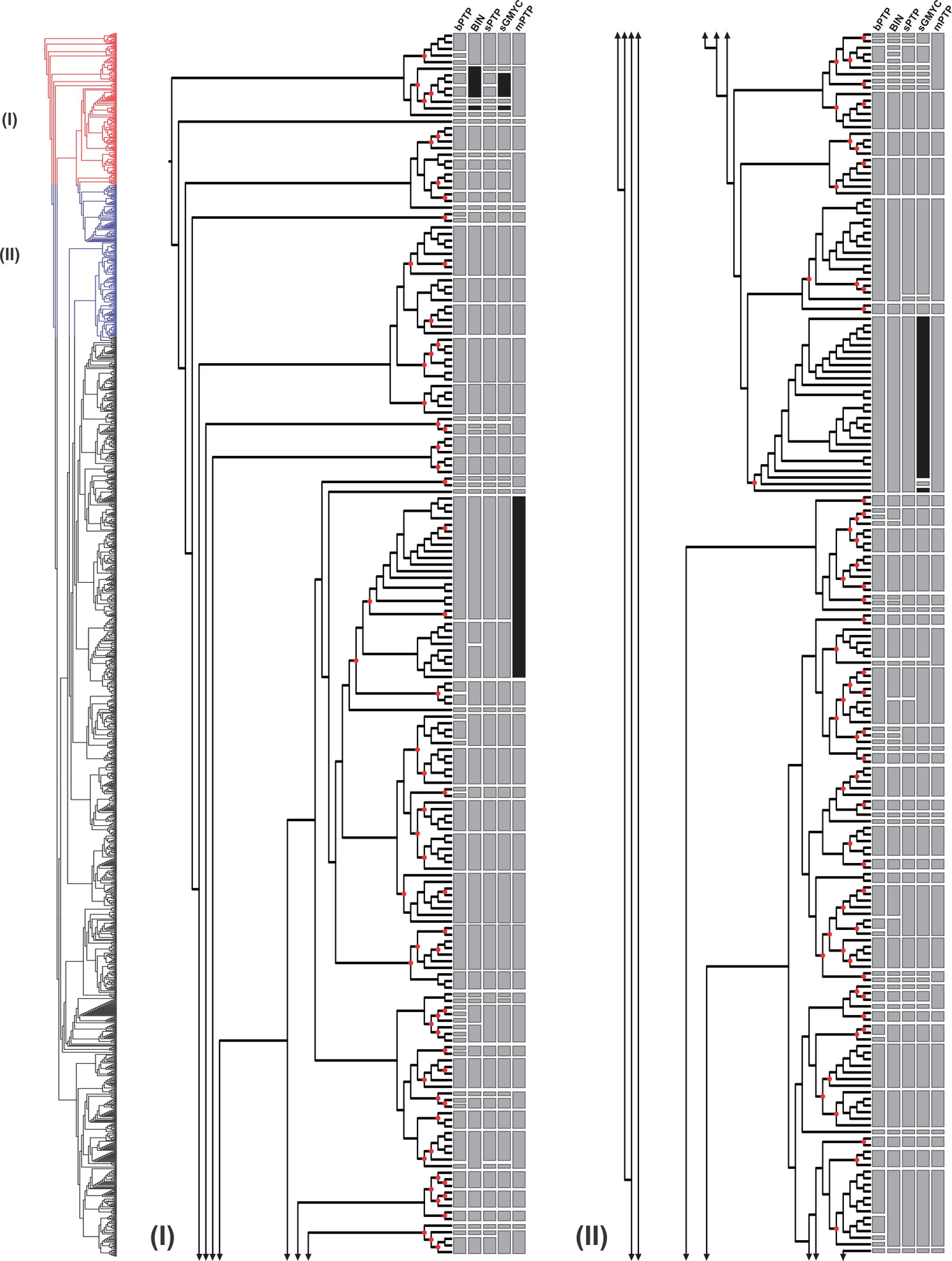
Aim The Neotropics, particularly South America, holds unparalleled high levels of species richness, when compared to other major biomes. Some neotropical areas are hotspots of a fragmentary known diversity of insects and are under manifest danger of biodiversity loss and climate change. Therefore, prompt estimates methods of its diversity are urgently required to complement slower traditional taxonomic approaches. Despite a variety of algorithms for delimiting species through single-locus DNA barcodes having been developed and applied for rapid estimates of species diversity in a wide array of taxa; however, tree-based and distance-based methods may lead to different group assignments, either overestimating or underestimating the number of putative species. Here, we investigate the performance of different DNA-based species delimitation approaches for a rapid biodiversity estimate of the diversity of Polypedilum (Chironomidae, Diptera) in South America. Location Worldwide Methods We analyze a mtDNA dataset comprising 1,492 specimens from 598 locations worldwide. Molecular operational taxonomic units (MOTUs) ranged from 267 to 520, based on the Barcode Index Number (BIN), Bayesian Poisson tree processes (bPTP), multi-rate Poisson tree processes (mPTP), single-rate Poisson tree processes (sPTP), and generalized mixed Yule coalescent (sGMYC) approaches. Results Our results highlight Polypedilum as a species-rich genus, yet incompletely documented, and found the sGMYC method to be the most adequate to estimate putative species in our dataset. Furthermore, based on these data, we describe the distribution of diversity and some biogeographical patterns of Polypedilum. Main Conclusions Findings imply the genus exhibited high levels of endemism and richness of species in the Neotropics, which confirmed our hypothesis that there are substantial differences in community structure between the Polypedilum fauna in South America and the neighboring regions.