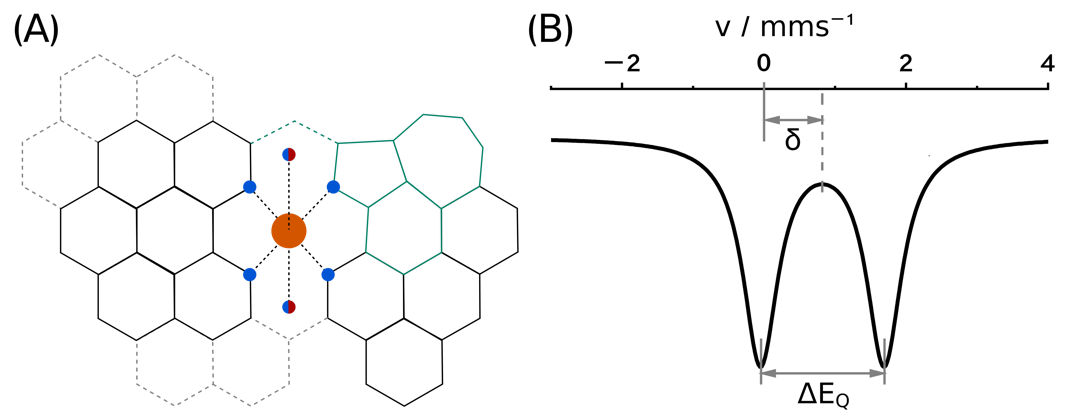
Single atom catalysts with iron ions in the active site, known as FeNC catalysts, show high activity for the oxygen reduction reaction and hence hold promise for access to low cost fuel cells. Due to the amorphous, multi-phase structure of the FeNC catalysts, the iron environment and its electronic structure are poorly understood. While it is widely accepted that the catalytically active site contains an iron ion ligated by several nitrogen donors embedded in a graphene-like plane, the exact structural details such as the presence or nature of axial ligands are unknown. Computational chemistry in combination with Mössbauer spectroscopy can help to unravel the geometric and electronic structures of the active sites. As a first step towards this goal, we present a calibration of computational Mössbauer spectroscopy for FeN4-like environments. The uncertainty of both the isomer shift and the quadrupole splitting prediction is determined, from which trust regions for the Mössbauer parameter predictions of computational FeNC models are derived. We find that TPSSh, B3LYP, and PBE0 perform equally well; the trust regions with B3LYP are 0.13 mm s−1 for the isomer shift and 0.45 mm s−1 for the quadrupole splitting. The calibration data is made publicly available in an interactive notebook that provides predicted Mössbauer parameters with individual uncertainty estimates from computed contact densities and quadrupole splitting values. We show that a differentiation of common FeNC Mössbauer signals by a separate analysis of isomer shift and quadrupole splitting will most likely be insufficient, whereas their simultaneous evaluation will allow the assignment to adequate computational FeNC models.