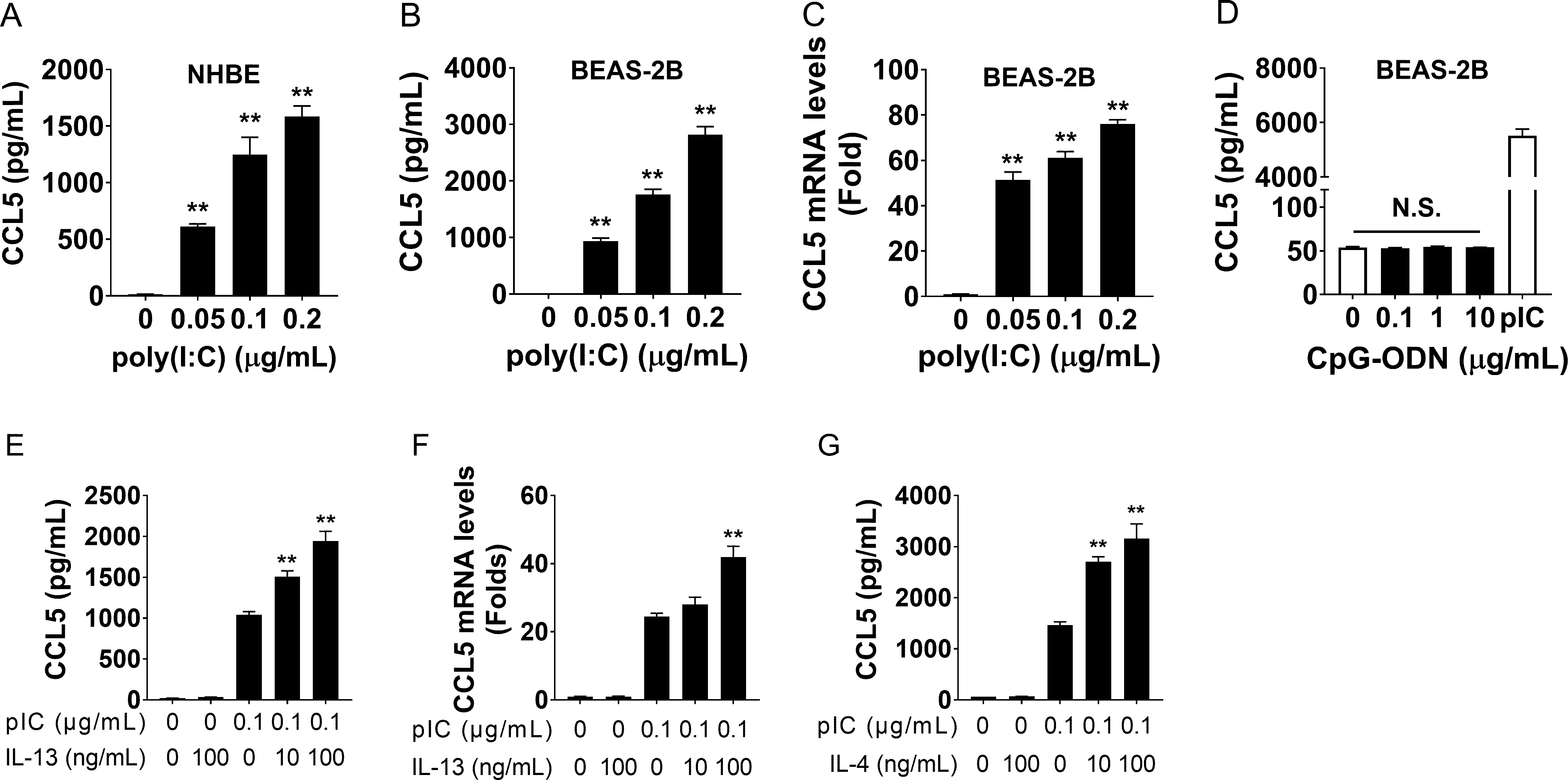
Rationale Severe eosinophilic asthma is characterized by airway eosinophilia and corticosteroid-resistance, commonly overlapping with type-2 inflammation. It has been reported that CCL5 is involved in asthma exacerbation due to RNA virus infections. We hypothesized that treatment with a virus-associated ligand and a Th2-cytokine can synergistically stimulate CCL5 production in bronchial epithelial cells. We also aimed to evaluate the mechanisms underlying CCL5 production in this in vitro model and to assess the potential of JAK1 as a novel therapeutic target via the use of ruxolitinib. Methods We stimulated primary normal human bronchial epithelial (NHBE) cells and BEAS-2B cells with poly (I:C) along with IL-13 or IL-4, and assessed CCL5 production. We also evaluated the signals involved in virus- and Th2-cytokine-induced CCL5 production and explored a therapeutic agent that attenuates the CCL5 production. Results Poly (I:C) stimulated NHBE and BEAS-2B cells to produce CCL5. Poly (I:C) and IL-13 increased CCL5 production. Poly (I:C)-induced CCL5 production occurred via the TLR3-IRF3 and IFNAR/JAK1-PI3K pathways, but not the IFNAR/JAK1-STATs pathway. In addition, IL-13 did not augment poly (I:C)-induced CCL5 production via the canonical IL-13R/IL-4R/JAK1-STAT6 pathway but likely via subsequent TLR3-IRF3-IFNAR/JAK1-PI3K pathways. JAK1 was identified to be a potential therapeutic target for severe eosinophilic asthma. The JAK1/2 inhibitor, ruxolitinib, was demonstrated to more effectively decrease CCL5 production in BEAS-2B cells than fluticasone propionate. Conclusion We have demonstrated that JAK1 is a possible therapeutic target for severe corticosteroid-resistant asthma with airway eosinophilia and persistent Th2-type inflammation, and that ruxolitinib has potential as an alternative pharmacotherapy.