AUTHOREA
Log in
Sign Up
Browse Preprints
LOG IN
SIGN UP
Paul Wilson
Professor
Trent University
Public Documents
8
September 12, 2023
Seq2Sat & SatAnalyzer toolkit: towards comprehensive microsatellite genotyping fr...
Peng Liu, Paul Wilson, Bridget Redquest, et al.
September 21, 2020
Considering Pleistocene North American wolves and coyotes in the eastern Canis origin...
Paul Wilson and Linda Y. Rutledge
September 02, 2020
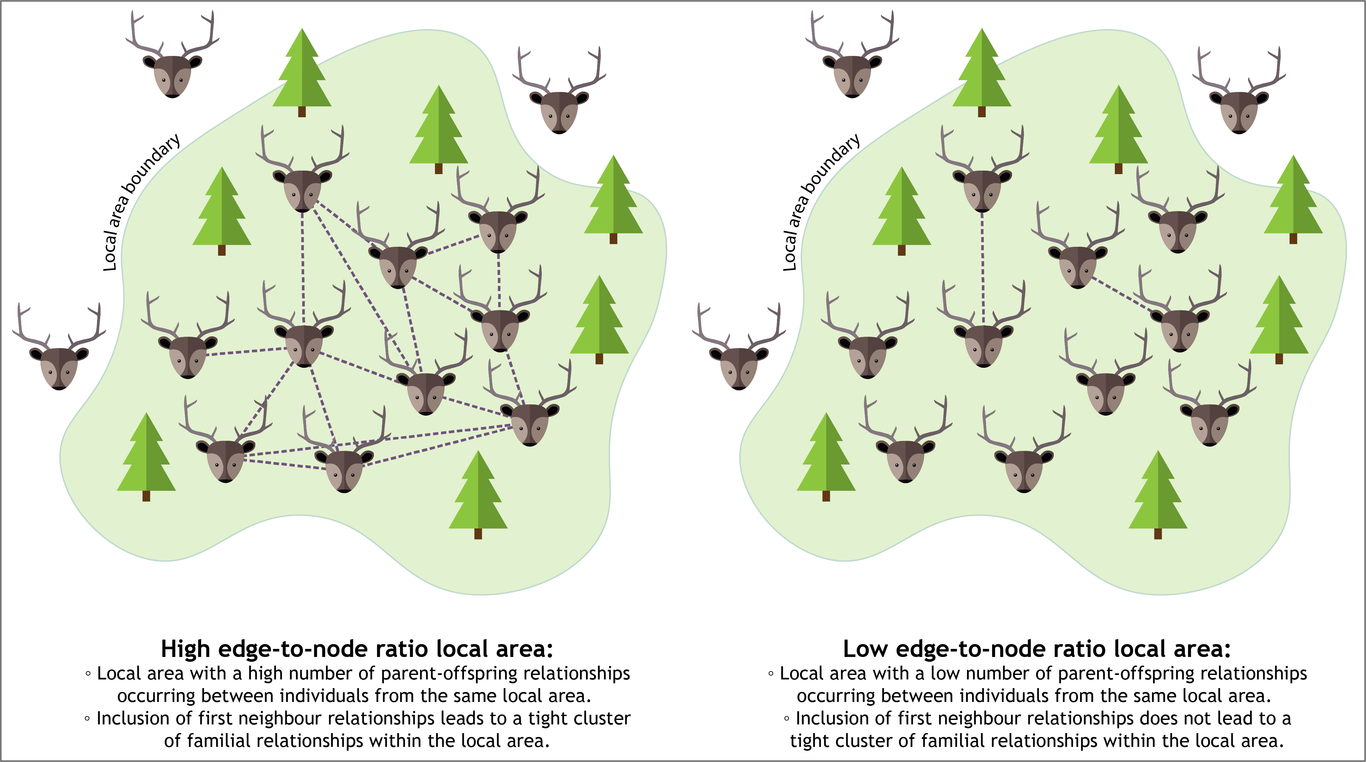
Spatial familial networks to infer demographic structure of wild populations
Samantha McFarlane, Micheline Manseau, Paul Wilson, et al.
August 23, 2020
Spatial and environmental influences on selection in a clock gene coding trinucleotid...
Melanie Prentice, Jeff Bowman, Dennis Murray, et al.
May 15, 2020
A framework for validating noninvasive genetic spatial capture-recapture studies for...
Samantha McFarlane, Micheline Manseau, Robin Steenweg, et al.
April 08, 2020
A framework for validating noninvasive genetic spatial capture-recapture studies for...
Samantha McFarlane, Micheline Manseau, Robin Steenweg, et al.
March 18, 2020
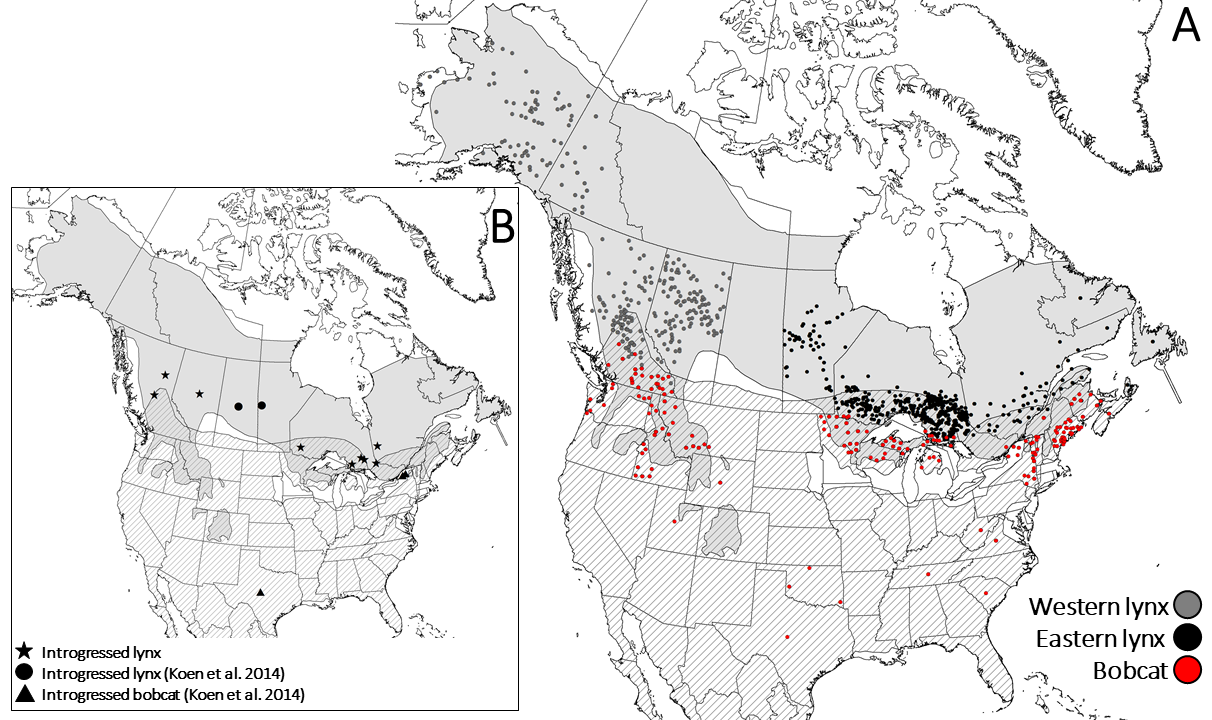
The role of introgression and ecotypic parallelism in delineating intra-specific cons...
Rebecca Taylor, Micheline Manseau, Rebekah Horn, et al.
April 28, 2020
Whole genome sequences from non-invasively collected samples
Rebecca Taylor, Micheline Manseau, Bridget Redquest, et al.