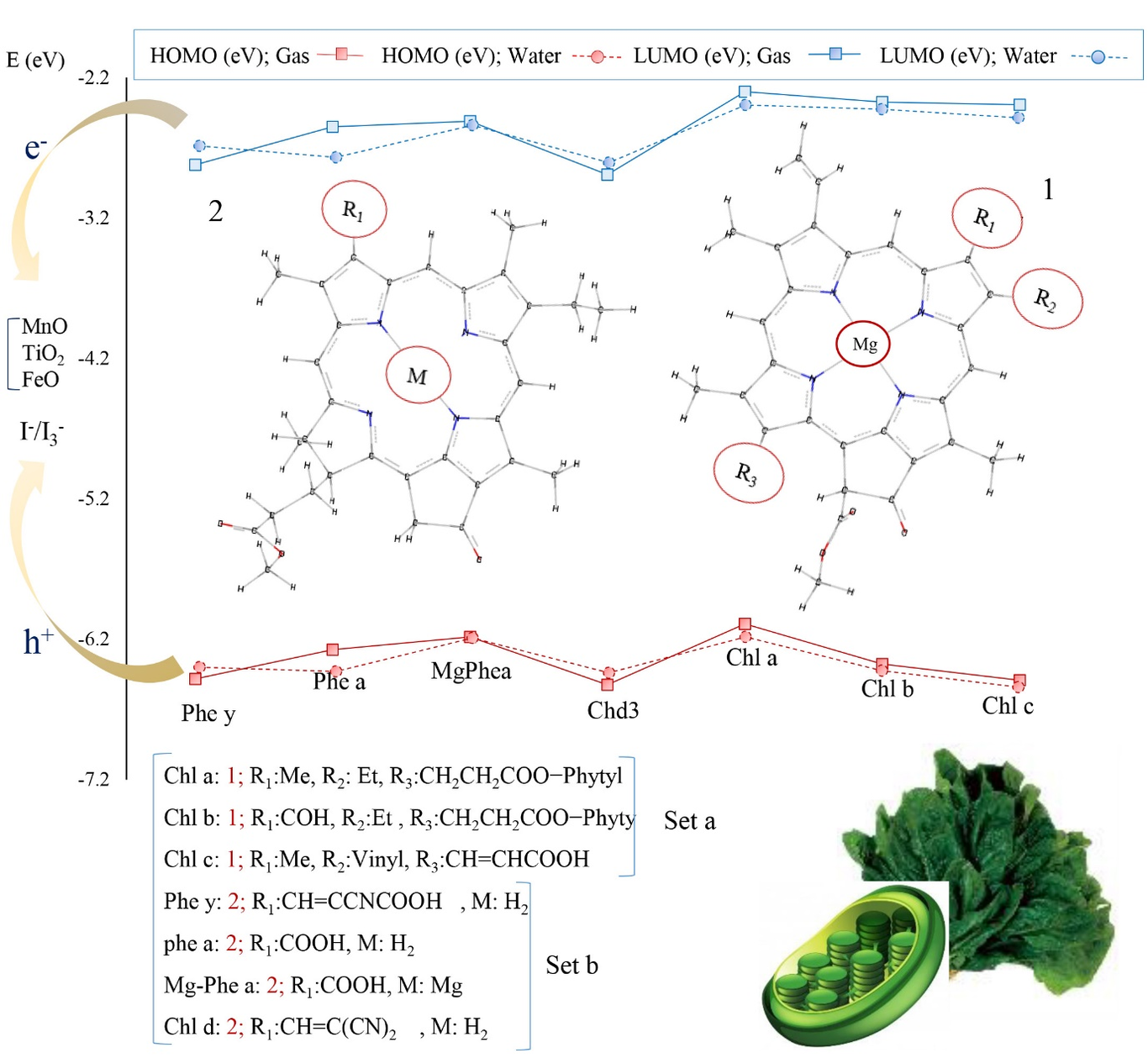
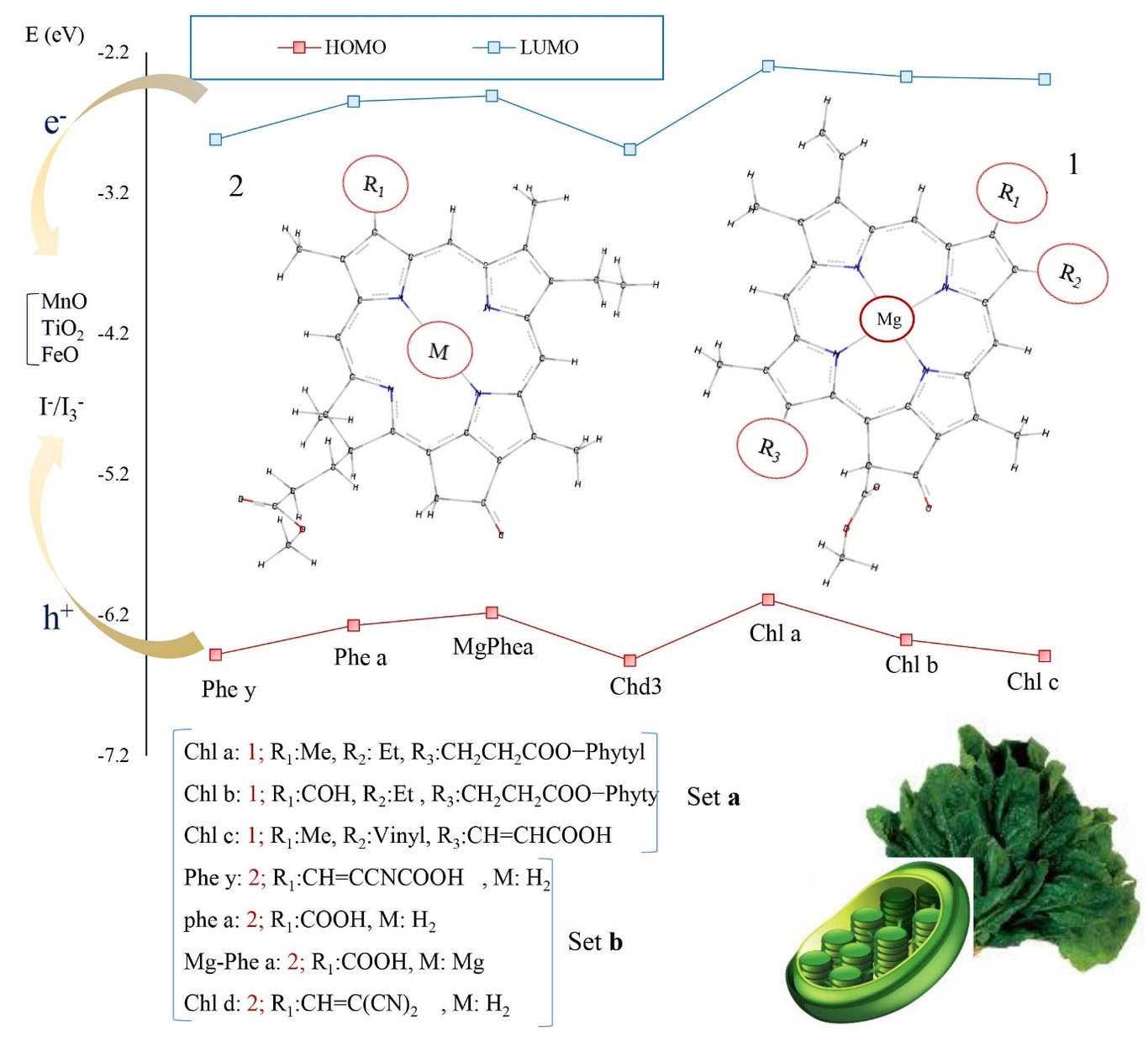
In this study, the photovoltaic properties of the organic dyes based on triphenylamine having a D--A structure including TC201, TC202, TC203, TC601, H-P, F-P, FF-P, T-F, and P1B were investigated theoretically. In this model, triphenylamine was used as an electron donor, cyanoacrylic acid, and benzoic acid as the electron acceptors, and anthracene phenyl, anthracene vinyl phenyl, anthracene ethynyl phenyl, ethynyl anthracene phenyl, styryl phenyl, styryl-2-fluorophenyl, styryl-2,6-difluorophenyl, styryl furan, and styryl as the π-conjugated systems. The results show that a change in the -conjugated system and electron acceptor affect the properties of the dye-sensitized solar cell (DSSC). Also, TC601 dye having the ethynyl anthracene phenyl -conjugated system shows the highest charge transfer distance (DCT) and the least overlap of the electron-hole distribution (S) in comparison with other dyes. Moreover, the presence of a triple bond in the vicinity of triphenylamine increases the resonance effect of the -electrons that facilitates the process of charge transfer in this dye. Spectroscopic analysis shows that H-P and F-P dyes have the higher molecular absorption coefficients and TC202, TC203, F-P, and T-F dyes show a red shift in comparison with other dyes. Moreover, the voltage-current curve of the studied dyes shows that the highest values of the open circuit voltage and short circuit current density are related to P1B and TC601 dyes, respectively. Finally, TC601 and P1B are proposed as the best candidates to be used in the DSSCs due to their maximum incident photon to current conversion efficiency.