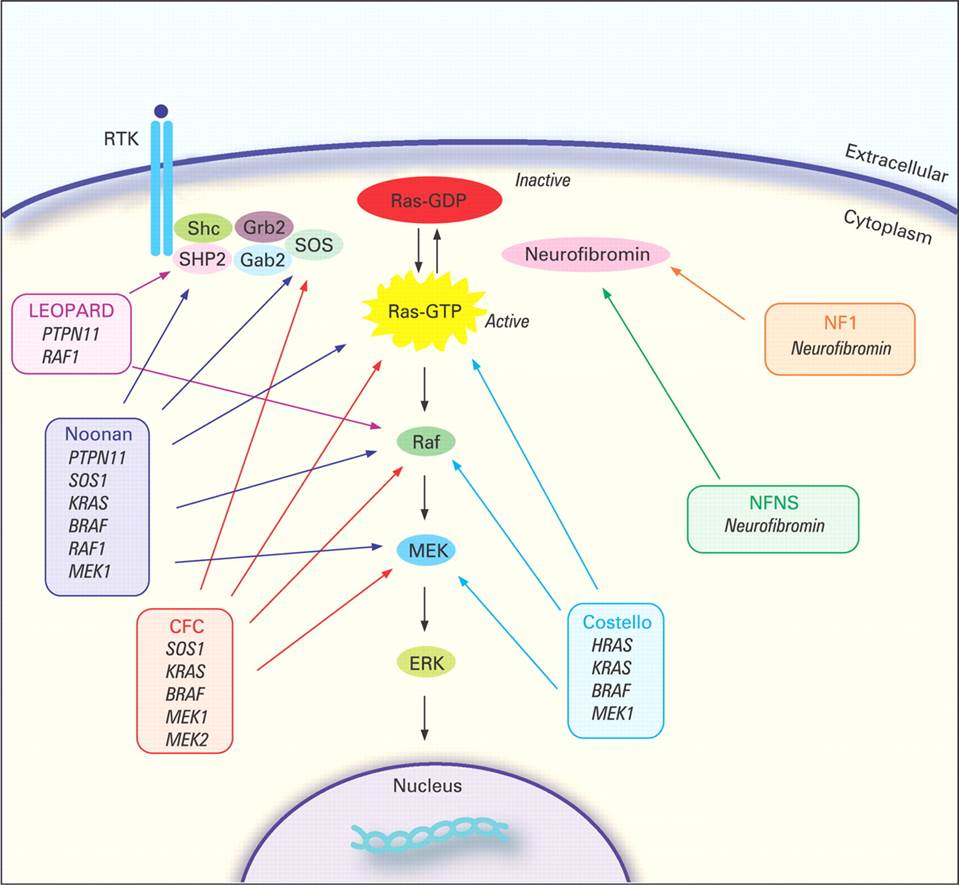
INTRODUCTION Sympathetic nervous system neural crest cholinergic localisation Neuroblastoma Overview Neuroblastoma (NB) is a pediatric cancer which arise from the sympathetic nervous system. It is the most common extracranial cancer of early childhood. The estimated incidence is of 1/8,000 to 1/10,000 births . NB arises from the sympathetic nervous system. Sixty-five percent of primary tumors occur within the abdomen, mostly in the adrenal medulla or sympathetic paraspinal ganglia. Other common locations are the neck when NB arise from the superior cervical ganglion (SCG), the thorax and the pelvis. Around 40% of patients present with a localized disease, others will present a metastatic disease. The most frequently observed metastatic sites are bone marrow, bone, lymph nodes, liver of subcutaneaous tissue. Among the metastatic patients, 10% have metastases in liver, skin or bone marrow that spontaneously regress. They compose the stage 4S (S=special) of the International Neuroblastoma Staging System (INSS) based on the anatomical presence of NB at diagnosis. The hallmark of neuroblastoma is its clinical heterogeneity whereas half of all cases classified as high-risk have an overall survival rate less than 40%, a subset of tumors will undergo spontaneous regressions. These tumors that regress spontaneously are more frequent than we expected. Autopsies of infants whose diagnosed cause of death was not cancer have shown an incidence of neuroblast precancer 40-fold higher than the incidence of the clinical disease . In addition, mass infant-screaning programs for the neuroblastoma tumor marker of catecholamines in urine samples found a two-fold higher incidence . In 1984, Shimada and colleagues classified tumors according to histopathological features. They were divided into 2 groups: stroma-poor and stroma-rich according to their organizational pattern (stromal development). These two groups were divided in subgroups associated with favorable or unfavorable cases. Predisposition Although the vast majority of NB cases are sporadic, rare familial and syndromic presentations are observed suggesting a genetic predisposition. Familial cases represent about 1% of all cases . The first predisposition mutation identified in neuroblastoma was in the paired-like homeobox 2b (_PHOX2B_) gene . This transcription factor promotes cell cycle exit and neuronal differentiation. Two types of mutations were identified in the _PHOX2B_ gene. PHOX2B protein harbors a homeodomain and 2 polyalanine stretches. Expansions of the second polyalanine stretch is often associated with congeital central hypoventilation syndrome (CCHS also refered as Ondine’s curse) alone whereas missense mutations or small insertions/deletions, non-polyalanine repeat expansion mutations (NPARMs), are associated with CCHS, NB, and Hirshprung’s disease. Hirshprung’s disease is due to a defect of migration of neural crest cells so it is characterized by the absence of the enteric ganglia in the distal part of the gastrointestinal tract. The patients are affected by chronic occlusions. About half of familial cases are explained by an activating mutation in the Anaplastic Lymphoma Kinase (_ALK_) gene. This will be described in details in the section [sectionALK]. The presence of constitutional _PHOX2B_ and _ALK_ mutations in familial neuroblastoma shows that these genes are involved in an early phase of tumor development that establishes susceptibility to NB. The RASopathies are developmental syndromes caused by germline mutations in genes that alter the Ras subfamily and Mitogen-activated protein kinases (MAPK) that control signal transduction (see figure [fig:FIGURE_RAS]). Few cases of NB were described in the familial tumor syndrome neurofibromatosis type 1 (NF1) well as in Noonan and Costello syndromes.