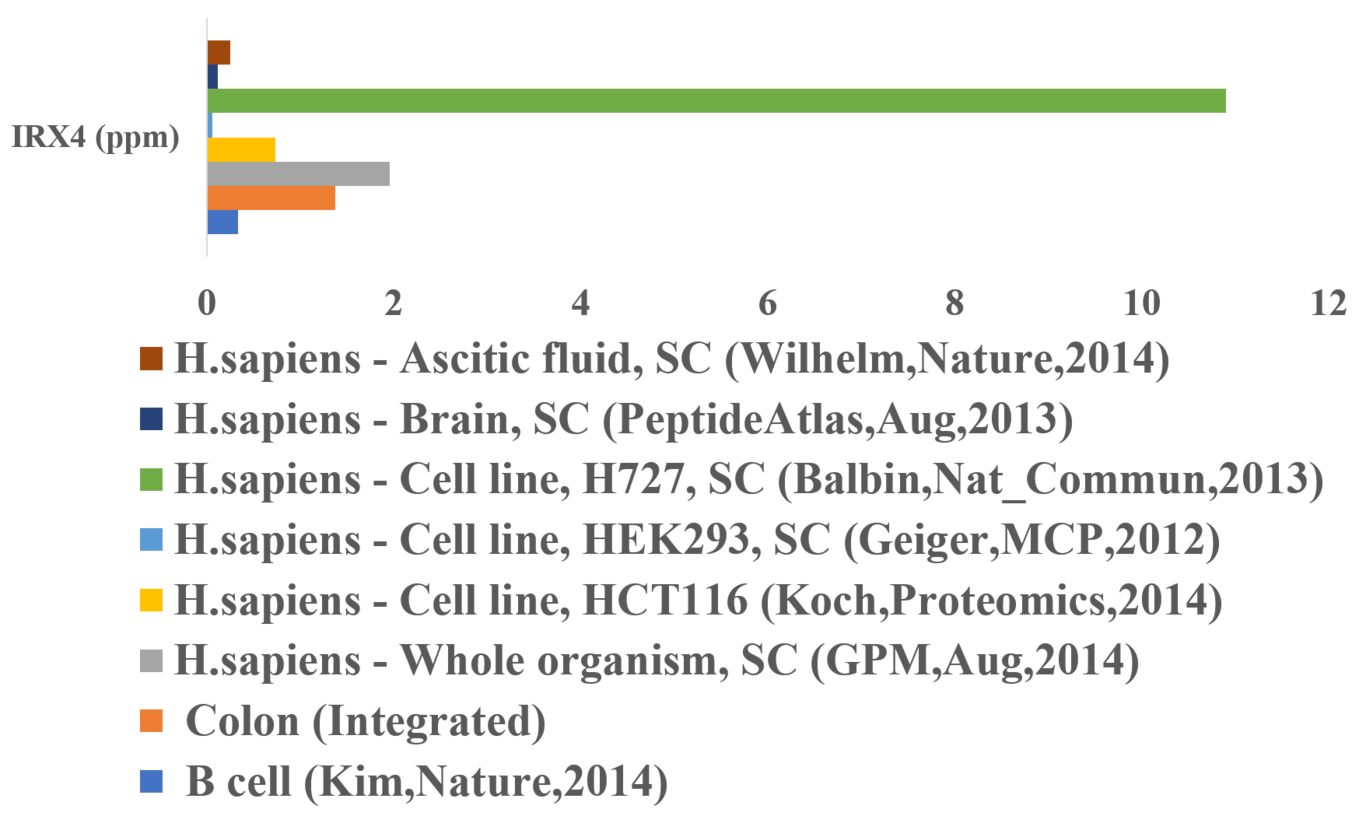
Iroquois Homeobox 4 (IRX4) belongs to a family of homeobox TFs having roles in embryogenesis, cell specification and organ development. Recently, Large scale Genome-Wide Association studies and epigenetic studies have highlighted the role of IRX4 and its associated variants in prostate cancer. No studies have investigated and characterized the structural aspect of the IRX4 homeodomain and its potential to bind to DNA. The current study uses sequence analysis, homology modelling and molecular dynamics simulations to explore IRX4 homeodomain-DNA recognition mechanisms and the role of somatic mutations affecting these interactions. Using publicly available databases, gene expression of IRX4 was found in different tissues, including prostate, heart, skin, vagina, and the protein expression was found in cancer cell lines (HCT166, HEK293), B cells, ascitic fluid and brain. Sequence conservation of the homeodomain shed light on the importance of N- and C-terminal residues involved in DNA binding. The specificity of IRX4 homodimer bound to consensus human DNA sequence was confirmed by molecular dynamics simulations, representing the role of conserved amino acids including R145, A194, N195, S190, R198 and R199 in binding to DNA. Additional N-terminal residues like T144 and G143 were also found to have specific interactions highlighting the importance of N-terminus of the homeodomain in DNA recognition. Additionally, the effects of somatic mutations, including the conserved Arginine (R145, R198 and R199) residues on DNA binding elucidated the importance of these residues in stabilizing the protein-DNA complex. Secondary structure and hydrogen bonding analysis showed the roles of specific residues (R145, T191, A194, N195, R198 and R199) in maintaining the homogeneity of the structure and its interaction with DNA. The differences in relative binding free energies of all the mutants shed light on the structural modularity of this protein and the dynamics behind protein-DNA interaction. We also have predicted that the C-terminal sequence of the IRX4 homeodomain could act as a potential cell-penetrating peptide, emphasizing the role these small peptides could play in targeting homeobox TFs.