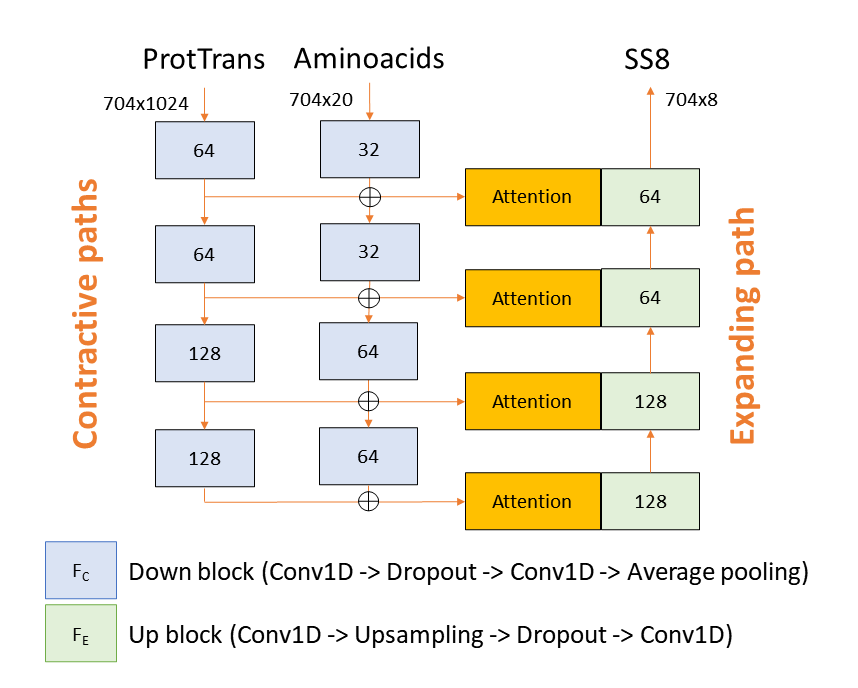
AlphaFold has transformed structure prediction by enabling highly accurate predictions on par with experimentally determined structures. Still, for difficult cases, in particular, multimers, there is still room for improvement. Important for the success of AlphaFold is its ability to assess its own predictions. The basic idea for the Wallner group in CASP15 was to exploit the excellent ranking score in AlphaFold by massive sampling. To this end, we ran AlphaFold using six different settings, with and without templates, and with an increased number of recycles using both multimer v1 and v2 weights. In all cases, the dropout layers were enabled at inference to sample the uncertainty and increase the diversity of the generated models. A median of 4,810 models per target was generated and almost all (35/38) received a ranking_confidence >0.7. Compared to other groups in CASP15, Wallner obtained the highest sum of Z-scores based on the DockQ score, 40.8 compared to 26.3 for the second highest, much higher than -0.2 achieved by the AlphaFold baseline method, NBIS-AF2-multimer. The improvement over the baseline is substantial with the mean DockQ increasing from 0.43 to 0.56, with several targets showing a DockQ score increase by +0.6 units. Remarkable, considering Wallner and NBIS-AF2-multimer were using identical input data. The reason for the success can be attributed to the diversified sampling using dropout with different settings and, in particular, the use of multimer v1, which seems to be much more susceptible to sampling compared to v2. The method is available here: http://wallnerlab.org/AFsample/.