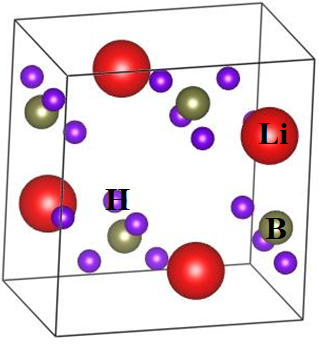
To cope with the energy crisis and global warming issues, researcher are rendering their efforts and paying their attentions to analyze and fabricate hydrogen storage devices. In this regard, we report a comprehensive study on the structural, vibrational, and optoelectronic properties of Lithium Borohydride (LiBH4), a hydrogen storage material. For this purpose, calculations of structural properties have been made using the local, non-local and hybrid functionals within the framework of density functional theory (DFT). The lattice constants for the orthorhombic phase are determined by applying LDA, PBE and HSE06 density functionals and their results are compared with available experimental and theoretical studies. In order to determine IR and Raman active modes of vibrations, vibrational spectroscopy has been utilized through Density Functional Perturbation Theory (DFPT) approach. Li, B and H atoms are noticed to be contributing in the modes of vibrations between different ranges of frequencies, i.e., 0 to 400 cm-1, 1100 to 1300 cm-1 and 2250 -2400 cm-1. The respective values of band gaps are found to be 6.35 eV, 6.81 eV and 7.58 eV for LDA, PBE and HSE06 functionals, respectively, leading to indicate insulating nature of LiBH4 which makes it a promising candidate for applications in optoelectronic devices. The mechanical analysis reveals that LiBH4 is a brittle material. The optical properties such as dielectric constant, refractive index, reflectivity, absorptivity, conductivity and loss function are also calculated with the aid of well-recognized relation of Kramer-Kronig. The plasma frequency is noted at the highest peak (13.7 eV) of the energy loss function.