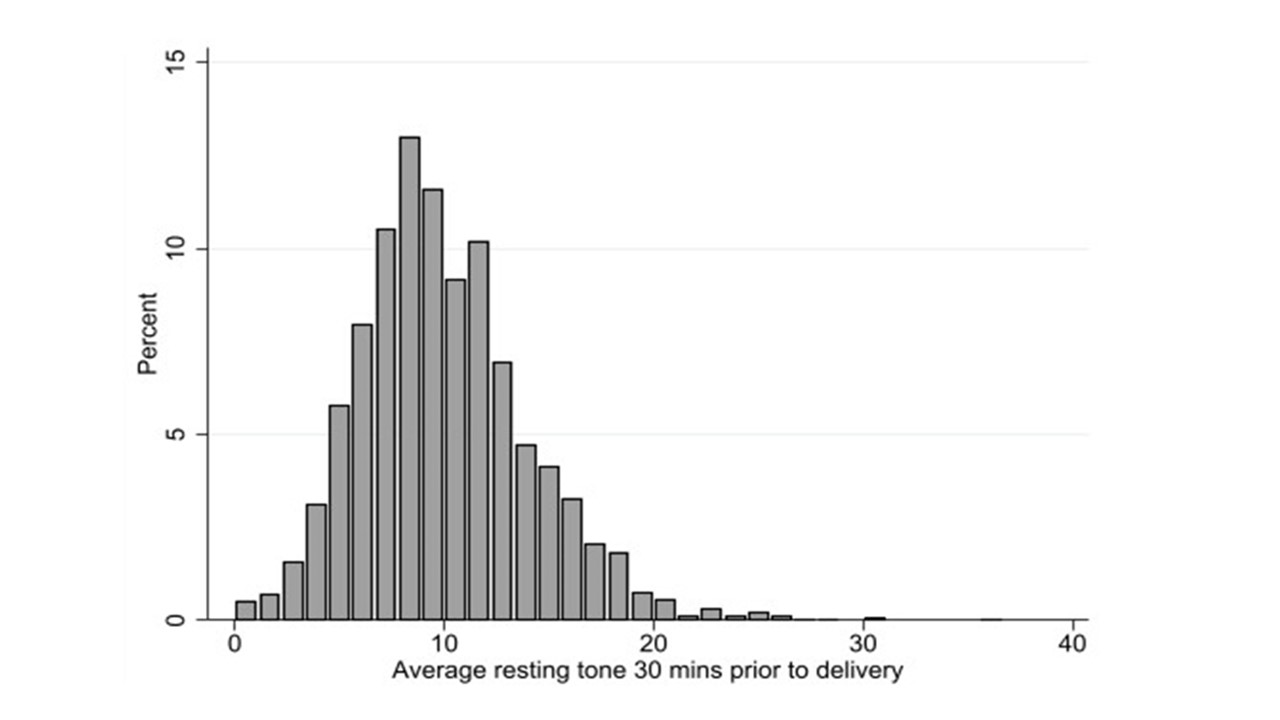
Objective: Internal contraction monitoring during the course of labor may identify elevated intrauterine resting tone. Our objective was to assess the association between elevated resting tone during labor and neonatal morbidity. Design: Secondary analysis of a prospective cohort study. Setting and Population: Term singleton patients with ruptured membranes and an intrauterine pressure catheter in place: Tertiary care hospital, United States of America Methods. Intrauterine resting tone was calculated as the average baseline pressure between contractions. The study group had elevated intrauterine resting tone, defined as intrauterine resting tone ≥75th percentile. Main Outcome Measures: Composite neonatal morbidity: hypoxic ischemic encephalopathy, hypothermia treatment, intubation, seizures, umbilical arterial pH ≥ 7.1, oxygen requirement, or death. Results: Of the 8580 patient in the cohort, 2210 (25.8%) were included. The median intrauterine resting tone was 9.7 mmHg (IQR 7.3-12.3 mmHg). Elevated resting tone was associated with shorter median duration of the first stage of labor (10.0 hrs vs 11.0 hrs, p <0.01) and lower rates of labor induction (p < 0.01). Neonatal composite morbidity was higher among patients with elevated intrauterine resting tone (5.1% vs 2.9%, p=0.01). After adjusting for chorioamnionitis and amnioinfusion, elevated intrauterine resting tone was associated with increased risk of neonatal morbidity (aOR 1.70, 95% CI 1.06-2.74). Compared to normal tone, elevated intrauterine resting tone was associated with mild acidemia and elevated lactate (aOR 1.81, 95% CI 1.38-2.37 and aOR 1.45, 95% CI 1.17-1.80, respectively). Conclusion: Elevated intrauterine resting tone is associated with increased risk of neonatal composite morbidity. Funding: None