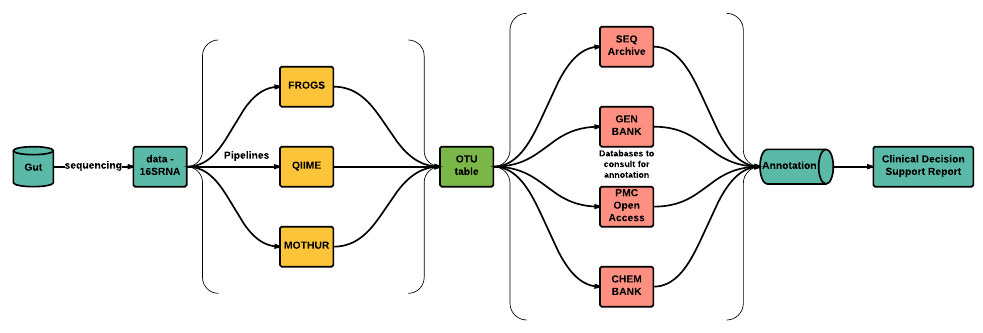
Chapter 1 (Rationale)IntroductionThe South African National Health Normative Standards Framework for Interoperability in eHealth, jointly written by the National Department of Health (NDoH) of South Africa and the Council for Scientific and Industrial Research (CSIR), is based on the existence of an standardized national electronic health record (EHR). This implies that there currently exists no standardized EHR in South Africa and thus no standard pertaining to the integration of Human Gut Microbiome (HGM) data or genomic data. This also attests a failure by the CSIR and NDoH to develop such standards. There are five different propriety health record systems in use (eHealth Strategy 2012, 2016) which allows for the creation of an EHR i.e., the EHR structure, across the country, are fragmented. This suggests that no international standardized EHR exists as the existence of such a document would not encourage fragmented EHRs. A lot of focus, by the CSIR and NDoH, has been put on interoperability, with respect to EHRs but no effort has been put into standards pertaining to the integration of genomic data into EHRs. Due to the lack of EHR standards it is difficult to develop clinical decision support tools to help clinicians at the point of care.Interoperability Standards in South AfricaAccording to the South African National Health Normative Standards Framework for Interoperability in eHealth the Health Level Seven (HL7) v2.x standard are currently in use throughout the country. HL7 v2.x is a popular messaging standard used to communicate eHealth data. It is further suggested in the document that the continuation of HL7 v2.x is to be used together with parts of HL7 v3 to achieve interoperability. The parts of HL7 v3 to be used are Clinical Care Document (CCD), Clinical Document Architecture (CDA) v2, and Consolidated Clinical Document Architecture (C-CDA). However, research has identified errors and permissible heterogeneity in C-CDA documents that will limit semantic interoperability (D’Amore et al., 2014). A new standard, Fast Health Interoperability Resource (FHIR), has been developed by HL7 and promises to be faster and easier to use than its predecessors HL7 v1, v2, and v3. In addition, FHIR is backwards compatible with previous HL7interoperability standards. The first version of FHIR was released in 2015 and thus was not considered in the South African National Health Normative Standards Framework for Interoperability in eHealth document. Current standards within healthcare is based on transferring entire documents instead of subsets of patient data. This means that physicians have to request an entire document and manually sift through it to find the information they would like to see whereas FHIR allows access to smaller ‘granular’ data elements. In addition, FHIR allows you to easily create custom clinical documents for any medical practitioner. For example, if a clinician wanted access to a patients name, gender, age,and ethnicity then only those elements will be sent to the clinician while with HL7 v1, v2, and v3, the clinician would receive a single document that contains all the patients information and would have to sift through it to get the sought after information. This process of sifting is both inefficient and does not scale.While there is no standardized format to add microbiome data into EHRs, we still need to establish how much microbiome data would needto be integrated. By analysing the description of microbiome pipelines of various bacterial species with varying levels of granularity we may identify and curate all necessary data elements needed to integrate into the EHR which in turn helps to develop a comprehensive microbiome annotation via the EHR. This annotation would help derive insights and inform decisions making, and tailor context specific treatment plans. Currently, researchers interpret microbiome data by searching additional data that will reveal function or search published literature that speaks to function of bacterial enzymes or association of bacteria with disease. For example, we may analyse microbiome data at varying levels of granularity through the use of pipelines such as FROGS (Find Rapidly OTUs with Galaxy Solution), MYcrobiota (an open-source, user-friendly galaxy application for microbiota determination and dynamic reporting from 16S sequences), or following the standard operating procedure for 16S rRNA diversity analysis as outlined by H3ABioNet to mention a few.Why Develop A Human Gut Microbiome Data Intergration StandardA HGM data integration standard would allow cohort-wide data mining of EHR databases and allow for micobiome-wide association studies and thus knowledge discovery. This enables context-specific and clinically actionable knowledge to be generated. Furthermore, biomedical informatics are moving towards the integration of genomic data into EHRs. The recent advancements of microbiome research plays an important role in the realization of personalized health care. The bacteria that colonize our colon contribute significantly to our physiology, nutritional well being, metabolic functions, the regulation of our immune system, and thought process (Rogers, 2015). For example, the gut microbiome has been linked to diseases such as obesity (Turnbaugh et al., 2006), circulatory disease (Holmes et al., 2008), inflammatory bowel disease Marchesi et al., 2007, and autism (Finegold, 2008). The gut microbiota has also been shown to influence drug metabolism and toxicity (Clayton et al., 2006), dietary calorific bio-availability (Hooper and Gordon, 2001), immune system conditioning and response (Macpherson et al., 2000), and post-surgical recovery (Kinross et al., 2011). Thus, microbiome data integration into EHRs is a crucial step towards enabling clinicians to better understand a patient’s response to treatment based on their microbiota and to enable them to create patient specific treatment plans.The Harvard Biomedical Informatics team has focused on a wide range of studies with regards to EHRs and patient care. These include identifying rheumatoid arthritis in EHRs via the use of a published phenotype algorithm, and revealing race-specific disease networks by a comparative analysis of population-scale phenomic data to mention a few. It is easy to see that the main goal of EHRs is to assist and support clinical care. In terms of research, this holds great promise as patient data now becomes a set of computable fine-grained longitudinal phenotypic profiles which may be used for knowledge discovery.How Microbiome Data Is GeneratedTo generate human gut microbiome data we follow a standard sequence workflow for microbiome research. Various microbiome studies typically have many elements in common, and a similar workflow. However, the particular questions being addressed will guide the experimental design and the methodology for generating, processing, and interpreting data (Jovel, 2016). The steps below illustrate a template for human gut microbiome studies. Firstly, sample collection is required so that DNA/RNA extraction may be done. Next, DNA/RNA extraction is done using sequencing platforms such as Illumina, PacBio, and Ion Torrent to name a few. After DNA/RNA extraction sequencing is done. The two main approaches for sequencing the microbiome are 16S ribosomal RNA (rRNA) gene amplicons and shotgun sequencing. The choice as to whether to use 16s or shotgun analysis for sequencing is dependent on the nature of the study being conducted. For a large number of samples. i.e., various patients or longitudinal studies etc, 16S is typically used whereas shotgun sequencing, while more expensive, offers increased resolution, enabling a more specific taxonomic and functional classification of sequences as well as the discovery of new bacterial genes and genomes (Jovel, 2016). Jovel asserts to explain the structure and function of the microbiome we should complementclinical and dietary information with proteomics, metabolomics, metatranscriptomics, and metadata i.e., in order to advance our understanding of the role the HGM plays in our health and its link to disease, integrative ‘omics’ techniques are required. Once sequencing is complete we typically do preprocessing of the sequences to eliminate errors etc from the sequences. If we wish we may or may not, after pre-processing, want to do sequence assembly and then carry out community characterization e.g., α-diversity and β-diversity. Below is a simplified version of the template described above.




/CodeCogsEqn (1).gif?1492726900)